Xenobiotics: Sources, Pathways, Degradation, and Risk Associated with Major Emphasis on Pharmaceutical Compounds
- First Online: 01 November 2023

Cite this chapter

- Manbir Singh 6 ,
- Ratish Chandra Mishra 6 ,
- Iqbal Shah 6 ,
- Vaishali Wadhwa 7 &
- Vikram Mor 8
177 Accesses
Xenobiotics are chemical substances which are alien or unnatural to the animal and human life. Xenobiotics include plant components, pharmaceutical drugs, pesticides, cosmetic products, added food flavors, fragrances, etc. At higher concentrations in environmental matrices, naturally occurring substances (endobiotics) may also be considered as xenobiotics. Xenobiotics are categorized as pesticides, pharmaceutical chemicals, personal care products, illicit narcotic drugs/substances, industrial/commercial goods, and nuclear waste and can be present in various environmental matrices. Xenobiotics are used by people and directly or indirectly penetrate in the different environmental matrices generating various metabolites and secondary products (some are even more toxic). Finally, plants, algae, and aquatic organisms take up xenobiotics leading to bioaccumulation, further causing biomagnification. One of the main obstacles to the sustainable water availability in urban systems is the presence of xenobiotics in aquatic ecosystems. In addition to the greater diversity of enzymes present in complex and varied community of microflora, the chemical distinctions between human and microbial transformations of ingested chemicals result from different selection processes that cause these activities. While host metabolism aids in the body’s elimination of xenobiotics, microbial changes to these substances and their human metabolites frequently promote microbial development by supplying nutrients or producing energy.
The amount of xenobiotics found in environmental matrices can be varied from ng/L to g/L. In both humans and animals, long-term chronic exposure to even little doses of xenobiotics may cause toxic, mutagenic, carcinogenic, or teratogenic effects. These compounds may block the enzyme’s active site or affect it in an allosteric way. Some xenobiotics including chlordecone, dichlorodiphenyltrichloroethane (DDT), and dichlorodiphenyldichloroethylene (DDE) show tendency to bioaccumulate, and even their low-level chronic exposures can potentially have an adverse impact on cell signaling pathways. In order to create safer molecules for use in human environment, knowledge of enhanced molecular designs may be useful along with mechanism of xenobiotics’ action. The following chapter explores types and sources of various xenobiotics, the introduction of xenobiotics in the atmosphere and soil, pathways and migration in the soil and aquatic systems (with emphasis on pharmaceutical chemicals), and decomposition of pharmaceutical chemicals in the environment.
This is a preview of subscription content, log in via an institution to check access.
Access this chapter
- Available as PDF
- Read on any device
- Instant download
- Own it forever
- Available as EPUB and PDF
- Durable hardcover edition
- Dispatched in 3 to 5 business days
- Free shipping worldwide - see info
Tax calculation will be finalised at checkout
Purchases are for personal use only
Institutional subscriptions
Similar content being viewed by others

Environmental Xenobiotics and Its Effects on Natural Ecosystem

Pollutants Biotransformation

Xenobiotics in the Food Chain: Quantitative Analysis, Toxic Impact, and Usage History
Ahel M, Molnar E, Ibric S et al (2000) Estrogenic metabolites of alkylphenol polyethoxylates in secondary sewage effluents and rivers. Water Sci Technol 42(7–8):15–22
Article CAS Google Scholar
Andreozzi R, Marotta R, Pinto G et al (2002) Carbamazepine in water: persistence in the environment, ozonation treatment and preliminary assessment on algal toxicity. Water Res 36(11):2869–2877
Armitage JM, Gobas FA (2007) A terrestrial food-chain bioaccumulation model for POPs. Environ Sci Technol 41(11):4019–4025
Barron L, Havel J, Purcell M et al (2009) Predicting sorption of pharmaceuticals and personal care products onto soil and digested sludge using artificial neural networks. Analyst 134(4):663–670
Benotti MJ, Trenholm RA, Vanderford BJ et al (2009) Pharmaceuticals and endocrine disrupting compounds in US drinking water. Environ Sci Technol 43(3):597–603
Bessa VS, Moreira IS, Tiritan ME et al (2017) Enrichment of bacterial strains for the biodegradation of diclofenac and carbamazepine from activated sludge. Int Biodet Biodegrad 120:135–142
Borgå K, Kidd KA, Muir DCG et al (2012) Trophic magnification factors: considerations of ecology, ecosystems, and study design. Integr Environ Assess Manag 8(1):64–84
Article Google Scholar
Borgman O, Chefetz B (2013) Combined effects of biosolids application and irrigation with reclaimed wastewater on transport of pharmaceutical compounds in arable soils. Water Res 47(10):3431–3443
Carter LJ, Harris E, Williams M et al (2014) Fate and uptake of pharmaceuticals in soil–plant systems. J Agric Food Chem 62(4):816–825
Clara M, Strenn B, Gans O et al (2005) Removal of selected pharmaceuticals, fragrances and endocrine disrupting compounds in a membrane bioreactor and conventional wastewater treatment plants. Water Res 39(19):4797–4807
Comber S, Gardner M, Sörme P (2018) Active pharmaceutical ingredients entering the aquatic environment from wastewater treatment works: a cause for concern. Sci Total Environ 613:538–547
Conder JM, Gobas FA, Borgå K et al (2012) Use of trophic magnification factors and related measures to characterize bioaccumulation potential of chemicals. Integrat Environ Ass Manag 8(1):85–97
De Oliveira M, Frihling BEF, Velasques J et al (2020) Pharmaceuticals residues and xenobiotics contaminants: occurrence, analytical techniques and sustainable alternatives for wastewater treatment. Sci Total Environ 705:135568
Delle Site A (2001) Factors affecting sorption of organic compounds in natural sorbent/water systems and sorption coefficients for selected pollutants: a review. J Phys Chem Ref Data 30(1):187–439
Ding J, Lu G, Liu J et al (2015) Evaluation of the potential for trophic transfer of roxithromycin along an experimental food chain. Environ Sci Pollut Res 22(14):10592–10600
Dodgen LK, Li J, Parker D et al (2013) Uptake and accumulation of four PPCP/EDCs in two leafy vegetables. Environ Poll 182:150–156
Ebele AJ, Abdallah MAE, Harrad S (2017) Pharmaceuticals and personal care products (PPCPs) in the freshwater aquatic environment. Emerg Contam 3(1):1–16
Embrandiri A, Kiyasudeen SK, Rupani PF et al (2016) Environmental xenobiotics and its effects on natural ecosystem. In: Plant responses to Xenobiotics. Springer, Singapore, pp 1–18
Google Scholar
Eur-Lex (2022) Available online: https://eur-lex.europa.eu/homepage.html . Accessed on 6 Sept 2022
European Medicines Agency (2022) Available online: https://www.ema.europa.eu/en . Accessed on 6 Sept 2022
Felton JS, Knize MG, Wu RW et al (2007) Mutagenic potency of food-derived heterocyclic amines. Mut Res/Fund Mol Mech Mutagen 616(1–2):90–94
Frank MP, Graebing P, Chib JS (2002) Effect of soil moisture and sample depth on pesticide photolysis. J Agric Food Chem 50(9):2607–2614
Franklin AM, Williams CF, Watson JE (2018) Assessment of soil to mitigate antibiotics in the environment due to release of wastewater treatment plant effluent. J Environ Qual 47(6):1347–1355
Fremlin KM, Elliott JE, Green DJ et al (2020) Trophic magnification of legacy persistent organic pollutants in an urban terrestrial food web. Sci Total Environ 714:136746
Gabet-Giraud V, Miege C, Choubert JM et al (2010) Occurrence and removal of estrogens and beta blockers by various processes in wastewater treatment plants. Sci Total Environ 408(19):4257–4269
Gao P, Mao D, Luo Y et al (2012) Occurrence of sulfonamide and tetracycline-resistant bacteria and resistance genes in aquaculture environment. Water Res 46(7):2355–2364
Geiler N (2006) PharmakaimAbwasserpfad. Wasser-&Abwassertechnik 7:13–15
Goldstein M, Shenker M, Chefetz B (2014) Insights into the uptake processes of wastewater-borne pharmaceuticals by vegetables. Environ Sci Technol 48(10):5593–5600
Gu C (2019) Urbanization: processes and driving forces. Sci China Earth Sci 62(9):1351–1360
Haham H, Oren A, Chefet B (2012) Insight into the role of dissolved organic matter in sorption of sulfapyridine by semiarid soils. Environ Sci Technol 46(21):11870–11877
Hamers T, Van den Berg JH, Van Gestel CA et al (2006) Risk assessment of metals and organic pollutants for herbivorous and carnivorous small mammal food chains in a polluted floodplain (Biesbosch, The Netherlands). Environ Poll 144(2):581–595
Heberer T (2002) Occurrence, fate, and assessment of polycyclic musk residues in the aquatic environment of urban areas—a review. Acta Hydrochim Hydrobio 30(5–6):227–243
Heemken OP, Reincke H, Stachel B et al (2001) The occurrence of xenoestrogens in the Elbe river and the North Sea. Chemosphere 45(3):245–259
Herklotz PA, Gurung P, Heuvel BV (2010) Uptake of human pharmaceuticals by plants grown under hydroponic conditions. Chemosphere 78(11):1416–1421
Karthigadevi G, Manikandan S, Karmegam N et al (2021) Chemico-nanotreatment methods for the removal of persistent organic pollutants and xenobiotics in water–a review. Bioresour Technol 324:124678
Kim S, Rossmassler K, Broeckling CD et al (2017) Impact of inoculum sources on biotransformation of pharmaceuticals and personal care products. Water Res 125:227–236
Kinne CA, Furlong ET, Werner SL et al (2006) Presence and distribution of wastewater-derived pharmaceuticals in soil irrigated with reclaimed water. Environ Toxicol Chem: Int J 25(2):317–326
Koba O, Golovko O, Kodešová R et al (2016) Transformation of atenolol, metoprolol, and carbamazepine in soils: the identification, quantification, and stability of the transformation products and further implications for the environment. Environ Poll 218:574–585
Kumar D, Chopra S (2020) Xenobiotic compounds in the environment: their fate, transport and removal. In: Proceedings of the 3rd National Conference on medical instrumentation, biomaterials and signal processing (NCMBS-20)
Levy SB (1998) The challenge of antibiotic resistance. Sci Am 278(3):32–39
Li WC (2014) Occurrence, sources, and fate of pharmaceuticals in aquatic environment and soil. Environ Poll 187:193–201
Li J, Dodgen L, Ye Q et al (2013) Degradation kinetics and metabolites of carbamazepine in soil. Environ Sci Technol 47(8):3678–3684
Lin AYC, Yu TH, Lateef SK (2009) Removal of pharmaceuticals in secondary wastewater treatment processes in Taiwan. J Haz Mat 167(1–3):1163–1169
Liu A, Patterson AD, Yang Z et al (2009) Fenofibrate metabolism in the cynomolgus monkey using ultraperformance liquid chromatography-quadrupole time-of-flight mass spectrometry-based metabolomics. Drug Metabo Disp 37(6):1157–1163
Loos R, Carvalho R, António DC et al (2013) EU-wide monitoring survey on emerging polar organic contaminants in wastewater treatment plant effluents. Water Res 47(17):6475–6487
Malchi T, Maor Y, Tadmor G et al (2014) Irrigation of root vegetables with treated wastewater: evaluating uptake of pharmaceuticals and the associated human health risks. Environ Sci Technol 48(16):9325–9333
Mansuy D (2013) Metabolism of xenobiotics: beneficial and adverse effects. Biol Aujourdhui 207(1):33–37
Massanyi P, Lukac N, Massanyi M et al (2020) Effects of Xenobiotics on animal reproduction in-vivo: Microscopical examination. Microsc Microanal 26(S1):63–63
Mathew BB, Singh H, Biju VG et al (2017) Classification, source, and effect of environmental pollutants and their biodegradation. J Environ Pathol Toxicol Oncol 36(1):55–71
Miller EL, Nason SL, Karthikeyan K et al (2016) Root uptake of pharmaceuticals and personal care product ingredients. Environ Sci Technol 50(2):525–554
Möller P, Morteani G, Dulski P (2003) Anomalous gadolinium, cerium, and yttrium contents in the adige and isarco river waters and in the water of their tributaries (Provinces Trento and Bolzano/Bozen, NE Italy). Acta Hydrochim Hydrobiol 31(3):225–239
Mompelat S, Le Bot B, Thomas O (2009) Occurrence and fate of pharmaceutical products and by-products, from resource to drinking water. Environ Int 35(5):803–814
Mountacer H, Atifi A, Wong-Wah-Chung P et al (2014) Degradation of the pesticide carbofuran on clay and soil surfaces upon sunlight exposure. Environ Sci Pollut Res 21(5):3443–3451
Narvaez VJF, Jimenez CC (2012) Pharmaceutical products in the environment: sources, effects and risks. Vitae 19(1):93–108
Nesterkova DV, Vorobeichik EL, Reznichenk IS (2014) The effect of heavy metals on the soil-earthworm-European mole food chain under the conditions of environmental pollution caused by the emissions of a copper smelting plant. Contemp Prob Ecol 7(5):587–596
Nikolaou A, Meric S, Fatta D (2007) Occurrence patterns of pharmaceuticals in water and wastewater environments. Analyt Bioanalyt Chem 387(4):1225–1234
Noman EA, Al-Gheethi AAS, Talip BA et al (2019) Xenobiotic organic compounds in greywater and environmental health impacts. In: Management of greywater in developing countries. Springer, Cham, pp 89–108
Chapter Google Scholar
Ofwat (2016) Water 2020: regulatory framework for wholesale markets and the 2019 price review. https://www.ofwat.gov.uk/wp-content/uploads/2015/12/pap_con20150912water2020.pdf
Olivieri AW, Seto E, Cooper RC et al (2014) Risk-based review of California’s water-recycling criteria for agricultural irrigation. J Environ Eng 140(6):04014015
Paltiel O, Fedorova G, Tadmor G et al (2016) Human exposure to wastewater-derived pharmaceuticals in fresh produce: a randomized controlled trial focusing on carbamazepine. Environ Sci Technol 50(8):4476–4482
Patel N, Khan MD, Shahane S et al (2020) Emerging pollutants in aquatic environment: source, effect, and challenges in biomonitoring and bioremediation-a review. Pollution 6(1):99–113
CAS Google Scholar
Patterson AD, Gonzalez FJ, Idle JR (2010) Xenobiotic metabolism: a view through the metabolometer. Chem Res Toxicol 23(5):851–860
Peck AM, Hornbuckle KC (2004) Synthetic musk fragrances in Lake Michigan. Environ Sci Technol 38(2):367–372
Peck AM, Linebaugh EK, Hornbuckle KC (2006) Synthetic musk fragrances in Lake Erie and Lake Ontario sediment cores. Environ Sci Technol 40(18):5629–5635
Pedersen JA, Yeager MA, Suffet IH (2003) Xenobiotic organic compounds in runoff from fields irrigated with treated wastewater. J Agri Food Chem 51(5):1360–1372
Piwowarska D, Kiedrzyńska E (2021) Xenobiotics as a contemporary threat to surface waters. Ecohydrol Hydrobiol 22(2):337–354
Pluth TB, Zanini LAG, Battisti IDE (2019) Pesticide exposure and cancer: an integrative literature review. Saúdeem Debate 43:906–924
Pollard KM, Christy JM, Cauvi DM et al (2018) Environmental xenobiotic exposure and autoimmunity. Curr Opin Toxicol 10:15–22
Rathore S, Varshney A, Mohan S et al (2022) An innovative approach of bioremediation in enzymatic degradation of xenobiotics. Biotechnol Gen Eng Rev 38(1):1–32
Ricking M, Schwarzbauer J, Franke S (2003) Molecular markers of anthropogenic activity in sediments of the Havel and Spree Rivers (Germany). Water Res 37(11):2607–2617
Roccaro P, Sgroi M, Vagliasindi FG (2013) Removal of xenobiotic compounds from wastewater for environment protection: treatment processes and costs. Chem Engg Transact 32:505–510
Sato T, Qadir M, Yamamoto S, Endo T et al (2013) Global, regional, and country level need for data on wastewater generation, treatment, and use. Agric Water Manag 130:1–13
Sauvêtre A, May R, Harpaintner R et al (2018) Metabolism of carbamazepine in plant roots and endophytic rhizobacteria isolated from Phragmites australis. J Haz Mater 342:85–95
Schirmer M, Strauch G, Reinstorf F, Schirmer K (2007) Urbane Hydrogeologie – Herausforderungen für Forschung und Praxis Grundwasser 12(3):178–188. https://doi.org/10.1007/s00767-007-0034-9
Singh R (2017) Biodegradation of xenobiotics-a way for environmental detoxification. Int J Dev Res 7(1):14082–14087
Sinha R (2002) An epidemiologic approach to studying heterocyclic amines. Mut Res/Fund Mol Mech Mut 506:197–204
Soucek P (2011) Xenobiotics. Encyclopedia of cancer, 3964–3967
Stachel B, Ehrhorn U, Heemken OP et al (2003) Xenoestrogens in the River Elbe and its tributaries. Environ Poll 124(3):497–507
Štefanac T, Grgas D, Landeka DT (2021) Xenobiotics—division and methods of detection: a review. J Xenobio 11(4):130–141
Stobierski T (2022) Globalization and the environment, Harvard Business School. Available online: https://online.hbs.edu/blog/post/globalization-effects-on-environment . Accessed on 6 Sept 2022
Sun J, Von Tungeln LS, Hines W et al (2009) Identification of metabolite profiles of the catechol-O-methyl transferase inhibitor tolcapone in rat urine using LC/MS-based metabonomics analysis. J Chromatograph B 877(24):2557–2565
Ternes TA (1998) Occurrence of drugs in German sewage treatment plants and rivers. Water Res 32(11):3245–3260
Ternes TA, Joss A, Siegrist H (2004) Peer reviewed: scrutinizing pharmaceuticals and personal care products in wastewater treatment. Environ Sci Technol 38(20):392A–399A
Thebo AL, Drechsel P, Lambin EF et al (2017) A global, spatially-explicit assessment of irrigated croplands influenced by urban wastewater flows. Environ Res Lett 12(7):074008
Thelusmond JR, Strathmann TJ, Cupples AM (2016) The identification of carbamazepine biodegrading phylotypes and phylotypes sensitive to carbamazepine exposure in two soil microbial communities. Sci Total Environ 571:1241–1252
Vieno N, Tuhkanen T, Kronberg L (2007) Elimination of pharmaceuticals in sewage treatment plants in Finland. Water Res 41(5):1001–1012
WHO (2022). https://www.who.int/news/item/04-04-2022-billions-of-people-still-breathe-unhealthy-air-new-who-data . Accessed on 6 Sept 2022
Windsor FM, Pereira MG, Tyler CR et al (2019) Persistent contaminants as potential constraints on the recovery of urban river food webs from gross pollution. Water Res 163:114858
Wu X, Conkle JL, Gan J (2012) Multi-residue determination of pharmaceutical and personal care products in vegetables. J Chromatograph A 1254:78–86
Yu X, Sui Q, Lyu S et al (2020) Municipal solid waste landfills: An underestimated source of pharmaceutical and personal care products in the water environment. Environment Sci Tech 54(16):9757–9768. https://doi.org/10.1021/acs.est.0c00565
Yang Y, Ok YS, Kim KH et al (2017) Occurrences and removal of pharmaceuticals and personal care products (PPCPs) in drinking water and water/sewage treatment plants: a review. Sci Total Environ 596:303–320
Zabrodskii PF (2020) The mechanisms of formation of Immunodeficiency’s, autoimmune reactions and hypersensitivity under the influence of Xenobiotics. J Immunol Aller 1:1–13
Zhang Y, Price GW, Jamieson R et al (2017) Sorption and desorption of selected non-steroidal anti-inflammatory drugs in an agricultural loam-textured soil. Chemosphere 174:628–637
Zhang C, Barron L, Sturzenbaum S (2021) The transportation, transformation and (bio) accumulation of pharmaceuticals in the terrestrial ecosystem. Sci Total Environ 781:146684
Zhu Y, Boye A, Body-Malapel M et al (2017) The toxic effects of xenobiotics on the health of humans and animals. Biomed Res Int 2017:4627872
Download references
Author information
Authors and affiliations.
Om Sterling Global University, Hisar, Haryana, India
Manbir Singh, Ratish Chandra Mishra & Iqbal Shah
Division of Soil and Crop Management, ICAR-CSSRI, Karnal, Haryana, India
Vaishali Wadhwa
SGT University, Gurugram, Haryana, India
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Manbir Singh .
Editor information
Editors and affiliations.
Department of Botany, Panjab University, Chandigarh, India
Rishikesh Singh
Department of Environmental Studies, PGDAV College, University of Delhi, Delhi, India
Pardeep Singh
Department of Botany, Deen Dayal Upadhyaya College, University of Delhi, Delhi, India
Sachchidanand Tripathi
Department of Forestry, Wildlife & Environmental Sciences, Guru Ghasidas Central University, Bilaspur, Chhattisgarh, India
K. K. Chandra
Department of Environmental Studies, Delhi College of Arts and Commerce University of Delhi, New Delhi, Delhi, India
Rahul Bhadouria
Rights and permissions
Reprints and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter
Singh, M., Mishra, R.C., Shah, I., Wadhwa, V., Mor, V. (2023). Xenobiotics: Sources, Pathways, Degradation, and Risk Associated with Major Emphasis on Pharmaceutical Compounds. In: Singh, R., Singh, P., Tripathi, S., Chandra, K.K., Bhadouria, R. (eds) Xenobiotics in Urban Ecosystems. Springer, Cham. https://doi.org/10.1007/978-3-031-35775-6_5
Download citation
DOI : https://doi.org/10.1007/978-3-031-35775-6_5
Published : 01 November 2023
Publisher Name : Springer, Cham
Print ISBN : 978-3-031-35774-9
Online ISBN : 978-3-031-35775-6
eBook Packages : Earth and Environmental Science Earth and Environmental Science (R0)
Share this chapter
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Publish with us
Policies and ethics
- Find a journal
- Track your research
- Search Menu
- Sign in through your institution
- Advance Articles
- High-Impact Collection
- Paper of the Year
- ToxSpotlight Articles
- Review Articles
- Author Guidelines
- Call for Papers
- Submission Site
- Open Access
- Early Career Researchers
- Why Publish with Us?
- About Toxicological Sciences
- About the Society of Toxicology
- Journals Career Network
- Editorial Board
- Advertising and Corporate Services
- Self-Archiving Policy
- Journals on Oxford Academic
- Books on Oxford Academic

Article Contents
Key issue no. 1: defining gut microbiomes associated with health, research need no. 1, key issue no. 2: defining causes and effects of altering the gut microbiome, research need no. 2, key issue no. 3: accounting for biotransformation, research need no. 3, key issue no. 4: determining biomarkers of disease and toxicity, research need no. 4, key issue no. 5: optimizing animal models, research need no. 5, recommendations, acknowledgments, the gut microbiome and xenobiotics: identifying knowledge gaps.
Vicki L. Sutherland and Charlene A. McQueen contributed equally to this study.
- Article contents
- Figures & tables
- Supplementary Data
Vicki L Sutherland, Charlene A McQueen, Donna Mendrick, Donna Gulezian, Carl Cerniglia, Steven Foley, Sam Forry, Sangeeta Khare, Xue Liang, Jose E Manautou, Donald Tweedie, Howard Young, Alexander V Alekseyenko, Frank Burns, Rod Dietert, Alan Wilson, Connie Chen, The Gut Microbiome and Xenobiotics: Identifying Knowledge Gaps, Toxicological Sciences , Volume 176, Issue 1, July 2020, Pages 1–10, https://doi.org/10.1093/toxsci/kfaa060
- Permissions Icon Permissions
There is an increasing awareness that the gut microbiome plays a critical role in human health and disease, but mechanistic insights are often lacking. In June 2018, the Health and Environmental Sciences Institute (HESI) held a workshop, “The Gut Microbiome: Markers of Human Health, Drug Efficacy and Xenobiotic Toxicity” ( https://hesiglobal.org/event/the-gut-microbiome-workshop ) to identify data gaps in determining how gut microbiome alterations may affect human health. Speakers and stakeholders from academia, government, and industry addressed multiple topics including the current science on the gut microbiome, endogenous and exogenous metabolites, biomarkers, and model systems. The workshop presentations and breakout group discussions formed the basis for identifying data gaps and research needs. Two critical issues that emerged were defining the microbial composition and function related to health and developing standards for models, methods and analysis in order to increase the ability to compare and replicate studies. A series of key recommendations were formulated to focus efforts to further understand host-microbiome interactions and the consequences of exposure to xenobiotics as well as identifying biomarkers of microbiome-associated disease and toxicity.
The gastrointestinal (GI) microbiota play an underlying role in health. Alterations in stability and functional capabilities of microbiota are associated with disease although is unclear whether this is a cause or a result of the disease. Although there is emerging research regarding the physiological functions of intestinal microbes, less is known about the consequences of chemically induced alterations to intestinal microbiome composition and toxicological effects to the host. The interactions of xenobiotics with the microbiota as a result of drug therapy or environmental exposures are of increasing interest to public health.
Health and Environmental Sciences Institute (HESI) assembled a cross-disciplinary group of experts ( Table 1 ) to examine gut microbial-host dynamics in order to better understand what is currently known about the effect of chemical exposures on the microbiome and to identify key knowledge gaps related to such exposures ( Figure 1 ).

Key data gaps in the microbiome research field identified at the 2018 HESI workshop. HESI’s June 2018 workshop identified key data gaps in the gut microbiome research field. The results identify needs for easily accessible biomarkers and improved human and animal experimental model studies for testing and validation of the impact of changes in the gut microbiome on human health outcomes.
Workshop Topics and Presenters
Experts from all sectors addressed state of the science and need for identification of biomarkers to advance understanding and decision making on efficacy and safety of xenobiotics. Presentations can be found on the HESI website here ( https://hesiglobal.org/event/the-gut-microbiome-workshop/ ).
The intestinal microbiome is made up of between 500 and 1000 bacterial species as well as viruses, archaea, and eukaryotic microorganisms, many of which play a role in human health and disease ( Backhed et al. , 2012 ; Gilbert et al. , 2018 ; Hanson and Weinstock, 2016 ; Ogilvie and Jones, 2015 ; Qin et al. , 2010 ; Tuddenham and Sears, 2015 ; Virgin, 2014 ; Ward et al. , 2018 ). There is great variation in species diversity, taxonomic composition, and population density of microbiota in the gut of humans and animal species. For those taxa that contribute to human health little is known about the part that each microbial species is filling, how well they perform that function, their interactions with other microorganisms and with the host, and where in the host they are acting ( Backhed et al. , 2012 ; Barratt et al. , 2017 ; Cho and Blaser, 2012 ; Gilbert et al. , 2018 ). Understanding the functions, features and normal ranges of the microbial communities that support health will be essential in addressing impacts of environmental agents and potentially identifying microbial configurations that result in disease ( Lloyd-Price et al. , 2016 , 2019 ). The inclusion of functional measurements may lead to different interpretations of microbiota diversity, compared with taxonomic classification alone, leading to greater analytical challenges ( Fu et al. , 2016 ; Zhu et al. , 2015 ). However, there is a growing consensus that functional characterizations are necessary for understanding and modeling the physiology of the microbiome ( Heintz-Buschart and Wilmes, 2018 ; Moya and Ferrer, 2016 ; Zhu et al. , 2015 ).
Before beneficial or adverse effects of chemical exposure can be determined, it is necessary to have a baseline for the bacterial species diversity as well as an understanding of the functional capacities associated with health. Given the considerable variation across human populations, among individuals and even within an individual, this will not be a single microbiome but a group of features, capabilities and characteristics of microbial communities that contribute to health. In defining such microbiomes, temporal variability in these communities, such as short-term perturbations associated with diet, lifestyle traits, antibiotic exposure, or acute illness, as well as more long-term impacts that may lead to deleterious chronic health outcomes must be considered ( Kundu et al. , 2017 ). In addition, factors such as geographic location, ethnicity, age, physical activity, genetics and gender, resistance (the ability to withstand stress or perturbations), and resilience (the capacity to return to a healthy state) influence an individual’s microbiome ( Backhed et al , 2012 ). For example, diet is associated with significant differences in microbiota both across populations and with longitudinal studies, yet there may not be apparent adverse impacts to human health ( Clemente et al , 2015 ; Yatsunenko et al , 2012 ). Such observations suggest that there may be functional redundancy in biochemical pathways in the microbiome ( Baumler and Sperandio, 2016 ). Large cohort studies may be needed in defining normal variations within a microbiome, especially if the studies include longitudinal sampling of the microbiome ( Faust et al. , 2015 ; Fettweis et al. 2019 ; Flores et al. , 2014 ; Lloyd-Price et al. , 2019 ; Proctor et al. , 2019 ; Vandeputte et al. , 2017a , b ; Yassour et al. , 2016 ; Zhou et al. , 2019 ). Measurement of blood metabolome is useful in predicting gut α-diversity in such studies ( Price et al. , 2017 ; Wilmanski et al. , 2019 ).
For most healthy adults, there appears to be a window of microbiome normalcy whose bounds are not typically exceeded, even though there may be minor transitory variation in composition observed with dietary changes or exposures to certain xenobiotics ( Kundu et al. , 2017 ; Moya and Ferrer, 2016 ). However, changes in microbial communities can be associated with adverse health consequences. In one model, this may occur if a person’s microbiome leaves their respective window of normalcy reaching a “tipping point” where their health is affected. Alternatively, rather than a distinct shift there can be stochastic changes in the microbiome leading to dispersion of the microbiome composition as well as increased functions that are related to disease ( Armour et al. , 2019 ; Zaneveld et al. 2017 ).
In order to understand, adverse health effects of the microbiome resulting from disease or xenobiotic exposure, there is a need to better understand the essential components, capabilities, and range of microbial variation linked to a person’s health. This requires comprehension of both the structure and the function of the microbial community. Identification of key conserved functional, metabolic, and biochemical pathways present in healthy individuals may provide a strong starting point.
It is evident that multiple factors contribute to the composition, diversity and function of the gut microbiome with regard to health, drug efficacy, and xenobiotic toxicity. The list includes genetics, diet, age, obesity, concomitant diseases, drugs, and gender to name a few. There is a need to not only understand the impact of a single factor on the dynamics between gut microbiome and the host but also to determine contributions of combinations of these factors. However, defining what constitutes microbial diversity and functionally of both healthy and unhealthy individuals will help to elucidate the relative contributions of host factors.
Large cohort studies are valuable sources of data for defining microbiomes associated with health, but comparison and reproducibility across such studies is hindered by differences in experimental design and data analysis. Variation and bias in sampling procedures, stabilization conditions, shipment and storage practices, methods for analyte extraction and purification, sample preparation and analysis, bioinformatic analysis algorithms as well as the reference databases used are contributing factors ( Backhed et al. , 2012 ; Gilbert et al. , 2018 ; Sinha et al. , 2017 ; Stulberg et al. , 2016 ). Standardization of methods and analytical approaches is needed to develop a well-characterized measurement pipeline. However, procedures may need to remain somewhat fluid to account for continuing technological developments and evolving understanding of the biology involved.
GI microbiota are essential in maintaining health and wellness by digesting food nutrients, producing endogenous metabolites and biotransforming xenobiotics ( Dietert and Silbergeld, 2015b ). Furthermore, in a healthy individual the gut microbiome, together with the intestinal mucosa, is necessary to maintain gut homeostasis and provide an epithelial barrier between the lumen and the rest of the body ( Hiippala et al. , 2018 ; Rogers, 2015 ).
Loss, disruption or dispersion of a functional microbiome can be associated with acute adverse health effects or chronic diseases. For example, antibiotics can alter a healthy individual’s microbiome resulting in diarrhea, leaving the body susceptible to the development of Clostridium difficile infection ( Backhed et al. , 2012 ; Schaffler and Breitruck, 2018 ; Schubert et al. , 2014 ). Changes in gut microbiota may be linked to the development of inflammatory bowel disease, colorectal cancer, and fatty liver disease ( Miyoshi et al. , 2017 ; Sharpton et al. , 2019 ; Wang et al. , 2017 ).
Many of these chronic diseases have inflammatory or altered immune system characteristics. Critical immune functions related to epithelial signaling, inflammatory responses, production of antimicrobial factors, and induction of Immunoglobulin A antibodies are propagated locally in the GI tract and associated secondary lymphoid tissues (Peyer’s Patches; Baumler and Sperandio, 2016 ; Dietert and Dietert, 2015a ; Hiippala et al. , 2018 ). The microbiome contributes to the development and maintenance of the immune system ( Agace and McCoy, 2017 ). Early life is characterized by a period of microbial flux and assembly which is affected by multiple factors such as manner of delivery (vaginal or c-section), nutrition (breast milk or formula) and antibiotic exposure. Although dynamic in nature, in adults the majority of the composition and function of the microbiome of a healthy person is fairly stable, but this decreases with age due to the loss of key species and a progressive gain of pathobiotic bacteria ( Buford, 2017 ).
The systemic influence of the GI microbiota on human health can be altered by exogenous compounds during drug therapy or environmental exposures. Antibiotics are used to remove a pathogen from the host, but an unintended consequence can be alteration of the composition and function of the gut microbiota ( Miyoshi et al. , 2017 ). Exposure to nicotine, arsenic, or polybrominated diphenyl ethers change the gut microbiome diversity ( Chi et al. , 2017 , 2018 ; Li et al. , 2018 ).
Exposure to xenobiotics can have effects in multiple ways. These include those that are transitory, established, or developmentally programmed ( Dietert and Dietert, 2015a ; Faust et al. , 2015 ; Fofanova et al. , 2016 ; Moya and Ferrer, 2016 ; Norman et al. , 2015 ; Pascal et al. , 2017 ; Sanz et al. , 2015 ; Stenman et al. , 2016 ). The transitory outcomes, such as changes after eating, are typically easily correctable and the gut microbiome return to the composition and functionality exhibited before the insult. Established outcomes occur when there is the alteration of a keystone species within the microbiota which would require significant host and microbiota changes to recover the original microbiota (eg, after C. difficile infection). The developmentally programmed outcomes are those that occur when the adverse impact is the result of alteration during a critical window of the microbiome development and can lead to later-in-life problems associated with the microbiome (eg, antibiotics during early childhood).
The gut microbiome plays an essential role in maintaining health and modification of the microbial communities can have negative effects in the body. Defining what changes in the microbiota are associated with acute or chronic adverse effects, as well as the magnitude of change necessary, is needed. Adverse effects can range from changes to the intestinal barrier integrity to alterations or dispersion of the normal microbiota of the host population of microbiota leading to a diseased state. Identification of such adverse effects may provide biomarkers of disease progression. To begin to identify causes and effects of changes in the microbial population, 2 approaches are possible. One is to start from a healthy individual’s baseline microbiome then determine deviations associated with a disease state. The second is determining what is a diseased state and ascertaining if there are key nodes, drivers, or contributing factors that move individuals from health to illness.
Despite the improvements in analytical, genomic, and bioinformatic techniques related to structure and function of the microbiome in health and disease, there is still a knowledge gap in the translation of high-throughput data from genotype to phenotype and microbiome composition stability/instability versus core function of the microbial community.
GI microbiota have the capacity to metabolize endogenous and exogenous compounds. The metabolites can have positive, neutral, or negative effects on the host. Endogenous microbial metabolites can exert physiological functions. Gut microbe-derived metabolites can signal via receptors at the epithelium interface and communicate with the host. For example, microbial metabolism of tryptophan generates aryl hydrocarbon receptor (AHR) ligands, such as indole, indirubin, and indigo ( Hubbard et al. , 2015 ). These ligands activate AHR and promote intestinal homeostasis through regulation of innate cytokine or chemokine gene expression, regulation of enterocyte differentiation, as well as regulation and development of intraepithelial lymphocytes and innate lymphoid cells. Gut microbial metabolism can result in products associated with adverse health and disease progression. The microbiome converts dietary lipid phosphatidylcholine to trimethylamine (TMA), which is then metabolized by hepatic flavin monooxygenases to TMA-N-oxide (TMAO). TMAO is a proatherogenic factor associated with cardiovascular disease (CVD; Brown and Hazen, 2015 ). Such findings suggest that endogenous microbial metabolites could be used as potential biomarkers of health or disease.
GI microbiota express biotransformation enzymes that metabolize a variety of therapeutic drugs and environmental compounds that can result in changes in efficacy and toxicity ( Klaassen and Kui, 2015 ; Spanogiannopoulos et al. , 2016 ). Gut bacteria can convert a prodrug to an active drug or detoxify the parent compound leading to modified drug availability, as well as changes in pharmacokinetics-pharmacodynamics (PKPDs). Sulfasalazine, a prodrug for ulcerative colitis, is directly metabolized by the gut bacteria at the azo bond to generate 2 bioactive metabolites, sulfapyridine (antimicrobial) and 5-aminosalicylate (anti-inflammatory; Peppercorn and Goldman, 1972 ). The cardiac drug glycoside digoxin undergoes reduction to an inactive metabolite, dihydrodigoxin, which is significantly decreased if antibiotics are preadministered ( Haiser et al. , 2013 ; Lindenbaum et al. , 1981 ). Microbial metabolites of a drug can also increase toxicity. The chemotherapeutic drug irinotecan is metabolized to SN-38 which is both the active and toxic metabolite. SN-38 is formed and glucuronidated by the liver and then transported to the gut where it undergoes deconjugation by gut bacteria. This results in longer gastrointestinal (GI) exposure, reabsorption, and greater bioavailability of SN-38 ( Wallace et al. , 2010 ). The gut microbiome is also implicated in the interindividual variability in metabolism of the analgesic and antipyretic acetaminophen. The endogenous microbial metabolite p-cresol and acetaminophen are substrates for sulfotransferases. Individuals with high levels of p-cresol generated by bacteria may have decreased acetaminophen metabolism through competition for hepatic sulfonation, which could result in an elevated risk for acetaminophen-induced hepatotoxicity ( Clayton et al. , 2009 ).
Nontherapeutic drugs and environmental xenobiotics can induce compositional changes and functional changes an in gut microbiota. In turn, these compounds may also be biotransformed by the microbiome. Arsenic is metabolized by gut bacteria and also alters the abundance and profile of the gut microbiome ( Chi et al. , 2018 ; Gokulan et al. , 2018 ; Lu et al. , 2014 ). Polychlorinated biphenyl or polybrominated diphenyl ether exposed mice show altered bile acid homeostasis resulting in part from changes in gut microbiota bile acid metabolism ( Cheng et al , 2018 ; Li et al , 2018 ). Gut anaerobes are capable of transforming Hg2 + to highly toxic and permeable methylmercury and significantly contributing to its body burden and poisoning ( Edwards and McBride, 1975 ).
In addition to directly biotransforming chemicals, the gut microbiome can influence the host xenobiotic metabolizing capability. Comparison of hepatic drug metabolizing genes in conventional and germ-free (GF) mice show that 34 genes, including Cyp3a, decrease, whereas 21 genes, including Cyp4a, increase ( Selwyn et al. , 2015 ). A cluster of 112 hepatic genes linked to xenobiotic metabolism and retinoid X receptor-inhibiting pathways are differentially expressed in conventional and GF mice. This results in more efficient pentobarbital metabolism and shorter anesthesia time in GF mice ( Bjorkholm et al. , 2009 ).
Current experimental approaches to evaluate microbe-mediated biotransformation primarily include: (1) in vitro incubation of individual bacterial strains, mixed cultures in bioreactors, or purified enzymes with compounds, (2) ex vivo incubation of a fecal microbiome community (or other GI regions of interest) with compounds, and (3) in vivo administration of compounds into animals such as rodents. These approaches provide evidence in support of microbial metabolism of many approved drugs ( Sousa et al. , 2008 ; Zimmermann et al. , 2019 ). However, more caution is required when extrapolating these results into humans than with traditional PKPD approaches, given the high diversity and interindividual variability of the human gut microbiome. Comprehensive and accurate modeling approaches for analyzing microbial metabolism information and incorporating into host PKPD models are necessary.
As more information on biotransformation of xenobiotics by the microbiome is published, a public database would be valuable. Such a database would be a repository centralizing comprehensive data that identifies compounds, responsible bacteria and/or enzymes, metabolites generated and metabolic pathways. This requires a collaborative effort involving researchers from academia, government and industry, working together with the regulatory agencies to promote a better understanding of microbiome-mediated biotransformation.
Biomarkers of disease and toxicity resulting from perturbations of the gut microbiota composition and function, from changes in microbial metabolites, or as a consequence of microbial biotransformation of xenobiotics would be useful. There are a number of characteristics for an ideal biomarker. It should be able to differentiate between disease progression and/or response to treatment. Stool samples or accessible tissues and/or biofluids should be used and analysis be affordable to facilitate acceptance and use for large scale screening. The biomarker needs to work for high-risk populations and distinguish between the disease and other potentially microbiome-mediated effects.
There are some examples of microbiome biomarkers of disease that are promising. Alterations in gut microbiota are associated with hypertension. Exposure of hypertensive rats to the antibiotic minocycline attenuates blood pressure ( Yang et al. , 2015 ). In animals and humans systolic blood pressure is correlated with microbiota composition and metabolites, as well as alterations in gut structure ( Kim et al. , 2018 ). Zonulin, a bacterial product and a marker of intestinal permeability, is elevated with high systolic blood pressure but is not an ideal biomarker since it is also associated with celiac disease ( Fasano et al. , 2000 ; Kim et al. , 2018 ). A more specific indicator may be the negative correlation of butyrate producing bacteria which alter systolic blood pressure and plasma butyrate levels ( Kim et al. , 2018 ).
TMAO blood levels are of interest as a biomarker for CVD, insulin resistance, and type 2 diabetes ( Miao, 2015 ). The production of TMAO in mammals primarily occurs via gut bacteria. Studies in humans and animals show circulating levels of TMAO are associated with risk and progression of CVD ( Tang et al. , 2014 ). Sensitive assays, particularly those amenable to the clinical setting, are being developed which will facilitate use of TMAO as a biomarker ( Garcia et al , 2017 ). The initial association of TMAO with CVD resulted from using untargeted metabolomics in cardiac patients ( Wang et al. , 2011 ). This approach can be expanded to use multiple data streams (clinical tests, metabolomes, proteomes, genome sequence, and microbiome) to develop correlation networks in order to identify bacterial analytes associated with normal physiology or disease ( Price et al. , 2017 ). For example, there is an association between phenylacetylglutamine and the Coriobacteriaceae and Mogibacteriaceae families. Phenylacetylglutamine is a microbial metabolite and is a risk factor for CVD in those with chronic kidney disease ( Poesen et al. , 2016 ).
Microbial biomarkers may also prove useful for identifying xenobiotic toxicity. Low dose exposure of mice to polychlorinated biphenyls alters microbiota composition leading to a greater abundance of species that generate secondary bile acids causing increases in serum bile acids ( Cheng et al. , 2018 ). At higher doses serum bile acids were not affected due to an increase in hepatic efflux transporters ( Cheng et al. , 2018 ).
Although in vivo studies are useful for discovering biomarkers, there is also a need for in vitro methods. In mice, tempol acts as an antioxidant by detoxifying reactive oxygen species and modulates the gut microbiome host signaling axis resulting in prevention of weight gain ( Cai et al. , 2016 ; Li et al ., 2013 ). Incubation of mouse cecal content with tempol and analysis of samples using flow cytometry as well as high-throughput mass spectrometry and nuclear magnetic resonance-based metabolomics show that tempol disrupted microbiota membrane physiology and metabolic activity consistent with the in vivo data ( Cai et al. , 2016 ). Such a multi-prong in vitro approach holds promise for screening xenobiotics for gut microbial toxicity and identifying potential biomarkers.
Case studies provide examples of potential biomarkers that could serve as the basis for noninvasive diagnostic tests to identify disease and track its progression. When an association between a disease and gut microbiota is recognized, additional work will be needed to distinguish between microbial changes that are a cause or a result of the disease. Biomarkers should also be applicable beyond their discovery cohorts and be able to distinguish between disease states of interest and nontarget diseases, as well as having a reasonable degree of specificity.
Discovery and qualification of biomarkers will also be advanced by the adoption of standard protocols which will generate robust and reproducible data that facilitates comparisons of studies. Detailed methods and procedures for analysis of gut microbiota structure and function that will be useful for toxicologic studies are available ( Nichols et al. , 2018 ).
Although animal species have limitations in completely replicating humans, they provide an opportunity to manipulate host genetics and the environment to allow insight into the relationship between the microbiota, xenobiotics, toxicity, and disease. Although species differences must be recognized in extrapolating animal studies to humans, such data are valuable.
Rodents are widely used in studying the microbiome. The compositions of gut microbiota vary among genetically distinct strains of mice ( Friswell et al. , 2010 ). Differences also occur in laboratory strains compared with genetically similar wild populations ( Rosshart et al. , 2017 ). GF mice and gene-knockout mice are also useful models. GF mice can be colonized with a single (mono-associated) agent, a defined bacterial combination such as altered Schardler flora or human fecal samples ( De Palma et al. , 2017 ; Franklin and Ericsson, 2017 ; Orcutt et al. , 1987 ; Ridaura et al. , 2013 ). Interestingly, introducing microbiota from wild mice into laboratory mouse strains increases resistance to influenza infection, decreases inflammation and increases resistance to certain types of cancer ( Rosshart et al. , 2017 ). The wild mouse microbiome develops as a result of evolutionary pressures due to continuous exposure to natural toxins and pathogens, thus it may offer a better model for human disease ( Rosshart et al. , 2017 ).
Disparities in gut microbiota of the same mouse strain can be attributed to vendor or institutional location ( Friswell et al. , 2010 ). Within a facility, the room, cage housing, food, water, and bedding all have an effect ( Ericsson et al. , 2018 ; Friswell et al. , 2010 ; Hildebrand et al. , 2013 ; Hugenholtz and de Vos, 2018 ; Robertson et al. , 2019 ).
Although rodents are the most commonly used models in microbiome studies, zebrafish are also proving to be useful. Zebrafish develop rapidly and externally to the mother, thereby enabling direct embryo exposures. The GI tract is homologous to that in mammals, although there are differences in the immune system. Zebrafish are kept at a lower temperature than mammals which may compromise the ability to study human-relevant microorganisms. Xenobiotic-microbiome interactions can be evaluated in the zebrafish model. For example, exposure of zebrafish larvae to the antimicrobial compound triclosan results in changes in the community structure of the microbiome. There is an increase in triclosan resistant species which results in a greater ability to biotransform triclosan ( Weitekemp et al. , 2019 ). Gut microbiota in zebrafish also alter metabolism and mediate the neurodevelopmental toxicity of 17-β-estradiol ( Catron et al. , 2019 ).
Study reproducibility in the biosciences is an ongoing concern ( Mullane and Williams, 2017 ). Variations in experimental design, methods, and statistical analysis can contribute to an inability to replicate results and conclusions. Complicating this further is the diversity and function of the gut microbiota ( Franklin and Ericsson, 2017 ; Turner, 2018 ).
Reproducibility in studies of gut microbiome host dynamics can be increased by recognizing the sources of variation, prioritizing them; then, identifying ways to address them ( Ericsson and Franklin, 2015 ; Franklin and Ericsson, 2017 ). To limit variation in mouse models, numerous approaches have been developed to standardize gut microbiota profiles including cohousing, cross fostering, and GF derivation, although each has advantages and disadvantages ( Ericsson and Franklin, 2015 ; Franklin and Ericsson, 2017 ). Maintaining a stable microbiome composition across animals and experiments is a problem that is not easily solved. Banking fecal samples annually and defining the microbiome at the initiation of a study would allow institutional drift to be monitored ( Franklin and Ericsson, 2017 ). In some instances, repeating studies in the second generation addresses the need for exposure to microbiota in early development ( Franklin and Ericsson, 2017 ). For zebrafish, protocols for GF derivation and gnotobiotic husbandry provide useful guidance for standardization ( Melancon et al. , 2017 ).
Often the discrepancies in reported results can be attributed to variations in study design. Unfortunately, key details are often missing from publications. Omissions can be reduced by adoption of Animal Research: Reporting of Experiments guidelines ( Kilkenny et al. , 2012 ). This checklist of 20 items covers species, strain and number of animals, husbandry, experimental design, methods, and statistical analysis.
Determining the optimal microbiota to facilitate translating results from mice to humans depends on the question being asked. The current model of colonizing GF mice with microbiota derived from inbred, wild murine, or human sources has great potential. This approach is currently limited by the availability of the number of defined combinations and characterized mouse or human material. The development of a wider array of standardized samples would be useful.
Several in vitro models of host-microbiome interactions have been described that complement in vivo studies ( Arnold et al. , 2016 ; von Martels et al. , 2017 ). Although there are anaerobic bacteria gut epithelial cell cocultures showing feasibility for experimental use, further refinements and standardization are still necessary ( von Martels et al. , 2017 ). It is also recognized that no one model is perfect, the best one to use will depend on the question being addressed.
The gut microbiome plays a pivotal part in health, but understanding its multiple roles still needs to be fully elucidated. HESI’s gut microbiome workshop was convened to identify research areas critical to determining how gut microbiome alterations may influence human health. Data gaps and research needs to address specific issues have been identified based on workshop presentations and breakout group discussions ( Table 2 ).
Key Issues and Research Needs
Key research areas and needs, as identified by workshop speakers and participants who participated in breakout group discussions.
There are 2 topics that cut across the key issues. The first is the lack of clarity in what constitutes gut microbiomes in healthy individuals. It is recognized that there are many factors that contribute to variation across and within human populations. Consequently, there is not a single microbiome but rather a suite of microbiomes that are associated with health. As yet, these have not yet been adequately defined. Having information on which species comprise such microbiomes and the normal range of variation is critical in order to define disease and adverse effects. The second is the need to adopt standards for methods, models and data analysis. This will facilitate comparisons across studies and reproducibility of data.
Moving forward the key recommendations are to focus efforts on the following important areas:
Defining the range of gut microbiota composition and function gut microbiomes associated with health and/or disease;
Identifying microbiome changes linked to disease and adverse health effects;
Characterizing the formation and function of microbiota metabolism of endogenous products on health and disease;
Determining the effects of xenobiotics on microbiota composition and function;
Increasing understanding of the impact of microbiota biotransformation of xenobiotics on efficacy and toxicity;
Identifying a suite of biomarkers to monitor health, disease and adverse effects resulting from microbiota-host interactions;
Standardizing variables such as husbandry, study design, sample collection, analysis, and statistical methods
Addressing these issues will provide further insight into the role of the microbiome in human health, disease and toxicity.
Health and Environmental Sciences Institute (HESI) is an independent nonprofit that collaboratively identifies and helps to resolve global and environmental challenges by convening experts through multi-sector forums. HESI’s work aims to move scientific principles into tested solutions that can be broadly applied to benefit health and the environment. The authors gratefully acknowledge the contributions of the speakers and the workshop participants, the discussions of which form the basis for this article. The views expressed in this article are those of the authors and do not necessarily reflect the views or policies of the author employers.
This work was provided in-kind by the Health and Environmental Sciences Institute (HESI) Microbiome Subcommittee, which is supported by sponsorships from member companies. HESI’s scientific initiatives are primarily supported by the in-kind contributions (from public and private sector participants) of time, expertise, and experimental effort. These contributions are supplemented by direct funding (that primarily supports program infrastructure and management) provided primarily by HESI’s corporate sponsors.
DECLARATION OF CONFLICTING INTERESTS
The authors of the article volunteered to serve on the HESI Microbiome committee and were involved in the planning and execution of the workshop. The views expressed in this article reflect the discussions from the workshop and do not necessarily reflect the views or policies of their employers.
Disclaimer: This workshop report is for information purposes only and shall not be construed to represent the official position or an obligation on the part of the U.S. Federal Government or any individual organization to provide support for any ideas or recommendations identified in it.
Agace W. W. , McCoy K. D. ( 2017 ). Regionalized development and maintenance of the intestinal adaptive immune landscape . Immunity 46 , 532 – 548 .
Google Scholar
Arnold J. W. , Roach J. , Azcarate-Peril M. A. ( 2016 ). Emerging technologies for gut microbiome research . Trends Microbiol. 24 , 887 – 901 .
Armour C. R. , Nayfach S. , Pollard K. S. , Sharpton T. J. ( 2019 ). A metagenomic meta-analysis reveals functional signatures of health and disease in the human gut microbiome . mSystems 4 , e00332 – 18 .
Backhed F. , Fraser C. M. , Ringel Y. , Sanders M. E. , Sartor R. B. , Sherman P. M. , Versalovic J. , Young V. , Finlay B. B. ( 2012 ). Defining a healthy human gut microbiome: Current concepts, future directions, and clinical applications . Cell Host Microbe 12 , 611 – 622 .
Barratt M. J. , Lebrilla C. , Shapiro H. Y. , Gordon J. I. ( 2017 ). The gut microbiota, food science, and human nutrition: A timely marriage . Cell Host Microbe 22 , 134 – 141 .
Baumler A. J. , Sperandio V. ( 2016 ). Interactions between the microbiota and pathogenic bacteria in the gut . Nature 535 , 85 – 93 .
Bjorkholm B. , Bok C. M. , Lundin A. , Rafter J. , Hibberd M. L. , Pettersson S. ( 2009 ). Intestinal microbiota regulate xenobiotic metabolism in the liver . PLoS One 4 , e6958 .
Brown J. M. , Hazen S. L. ( 2015 ). The gut microbial endocrine organ: Bacterially derived signals driving cardiometabolic diseases . Annu. Rev. Med. 66 , 343 – 359 .
Buford T. W. ( 2017 ). (Dis)Trust your gut: The gut microbiome in age-related inflammation, health, and disease . Microbiome 5 , 80 .
Cai J. , Zhang L. , Jones R. A. , Correll J. B. , Hatzakis E. , Smith P. B. , Gonzalez F. J. , Patterson A. D. ( 2016 ). Antioxidant drug tempol promotes functional metabolic changes in the gut microbiota . J. Proteome Res. 15 , 563 – 571 .
Catron T. R. , Swank A. , Wehmas L. C. , Phelps D. , Keely S. P. , Brinkman N. E. , McCord J. , Singh R. , Sobus J. , Wood C. E. , et al. . ( 2019 ). Microbiota alter metabolism and mediate neurodevelopmental toxicity of 17beta-estradiol . Sci. Rep. 9 , 7064 .
Cheng S. L. , Li X. , Lehmler H. J. , Phillips B. , Shen D. , Cui J. Y. ( 2018 ). Gut microbiota modulates interactions between polychlorinated biphenyls and bile acid homeostasis . Toxicol. Sci. 166 , 269 – 287 .
Chi L. , Gao B. , Tu P. , Liu C. W. , Xue J. , Lai Y. , Ru H. , Lu K. ( 2018 ). Individual susceptibility to arsenic-induced diseases: The role of host genetics, nutritional status, and the gut microbiome . Mamm. Genome 29 , 63 – 79 .
Chi L. , Mahbub R. , Gao B. , Bian X. , Tu P. , Ru H. , Lu K. ( 2017 ). Nicotine alters the gut microbiome and metabolites of gut-brain interactions in a sex-specific manner . Chem. Res. Toxicol. 30 , 2110 – 2119 .
Cho I. , Blaser M. J. ( 2012 ). The human microbiome: At the interface of health and disease . Nat. Rev. Genet. 13 , 260 – 270 .
Clayton T. A. , Baker D. , Lindon J. C. , Everett J. R. , Nicholson J. K. ( 2009 ). Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism . Proc. Natl. Acad. Sci. U.S.A. 106 , 14728 – 14733 .
Clemente J. C. , Pehrsson E. C. , Blaser M. J. , Sandhu K. , Gao Z. , Wang B. , Magris M. , Hidalgo G. , Contreras M. , Noya-Alarcón Ó. , et al. . ( 2015 ). The microbiome of uncontacted Amerindians . Sci. Adv . 1 , e1500183 .
De Palma G. , Lynch M. D. , Lu J. , Dang V. T. , Deng Y. , Jury J. , Umeh G. , Miranda P. M. , Pigrau Pastor M. , Sidani S. , et al. . ( 2017 ). Transplantation of fecal microbiota from patients with irritable bowel syndrome alters gut function and behavior in recipient mice . Sci. Transl. Med. 9 , eaaf6397 .
Dietert R. R. , Dietert J. M. ( 2015 a). The microbiome and sustainable healthcare . Healthcare (Basel) 3 , 100 – 129 .
Dietert R. R. , Silbergeld E. K. ( 2015 b). Biomarkers for the 21st century: Listening to the microbiome . Toxicol. Sci. 144 , 208 – 216 .
Edwards T. , McBride B. C. ( 1975 ). Biosynthesis and degradation of methylmercury in human faeces . Nature 253 , 462 – 464 .
Ericsson A. C. , Franklin C. L. ( 2015 ). Manipulating the gut microbiota: Methods and challenges . ILAR J. 56 , 205 – 217 .
Ericsson A. C. , Gagliardi J. , Bouhan D. , Spollen W. G. , Givan S. A. , Franklin C. L. ( 2018 ). The influence of caging, bedding, and diet on the composition of the microbiota in different regions of the mouse gut . Sci. Rep. 8 , 4065 .
Fasano A. , Not T. , Wang W. , Uzzau S. , Berti I. , Tommasini A. , Goldblum S. E. ( 2000 ). Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease . Lancet 355 , 1518 – 1519 .
Faust K. , Lahti L. , Gonze D. , de Vos W. M. , Raes J. ( 2015 ). Metagenomics meets time series analysis: Unraveling microbial community dynamics . Curr. Opin. Microbiol. 25 , 56 – 66 .
Fettweis J. M. , Serrano M. G. , Brooks J. P. , Edwards D. J. , Girerd P. H. , Parikh H. I. , Huang B. , Arodz T. J. , Edupuganti L. , Glascock A. L. , et al. . ( 2019 ). The vaginal microbiome and preterm birth . Nat. Med. 25 , 1012 – 1021 .
Flores G. E. , Caporaso J. G. , Henley J. B. , Rideout J. R. , Domogala D. , Chase J. , Leff J. W. , Vazquez-Baeza Y. , Gonzalez A. , Knight R. , et al. . ( 2014 ). Temporal variability is a personalized feature of the human microbiome . Genome Biol. 15 , 531 .
Fofanova T. Y. , Petrosino J. F. , Kellermayer R. ( 2016 ). Microbiome-epigenome interactions and the environmental origins of inflammatory bowel diseases . J. Pediatr. Gastroenterol. Nutr. 62 , 208 – 219 .
Franklin C. L. , Ericsson A. C. ( 2017 ). Microbiota and reproducibility of rodent models . Lab. Anim. (NY ) 46 , 114 – 122 .
Friswell M. K. , Gika H. , Stratford I. J. , Theodoridis G. , Telfer B. , Wilson I. D. , McBain A. J. ( 2010 ). Site and strain-specific variation in gut microbiota profiles and metabolism in experimental mice . PLoS One 5 , e8584 .
Fu B. C. , Randolph T. W. , Lim U. , Monroe K. R. , Cheng I. , Wilkens L. R. , Le Marchand L. , Hullar M. A. , Lampe J. W. ( 2016 ). Characterization of the gut microbiome in epidemiologic studies: The multiethnic cohort experience . Ann. Epidemiol. 26 , 373 – 379 .
Garcia E. , Wolak-Dinsmore J. , Wang Z. , Li X. S. , Bennett D. W. , Connelly M. A. , Otvos J. D. , Hazen S. L. , Jeyarajah E. J. ( 2017 ). NMR quantification of trimethylamine-N-oxide in human serum and plasma in the clinical laboratory setting . Clin. Biochem . 50 , 947 – 955 .
Gilbert J. A. , Blaser M. J. , Caporaso J. G. , Jansson J. K. , Lynch S. V. , Knight R. ( 2018 ). Current understanding of the human microbiome . Nat. Med. 24 , 392 – 400 .
Gokulan K. , Arnold M. G. , Jensen J. , Vanlandingham M. , Twaddle N. C. , Doerge D. R. , Cerniglia C. E. , Khare S. ( 2018 ). Exposure to arsenite in CD-1 mice during juvenile and adult stages: Effects on intestinal microbiota and gut-associated immune status . mBio 9 ,
Haiser H. J. , Gootenberg D. B. , Chatman K. , Sirasani G. , Balskus E. P. , Turnbaugh P. J. ( 2013 ). Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta . Science 341 , 295 – 298 .
Hanson B. M. , Weinstock G. M. ( 2016 ). The importance of the microbiome in epidemiologic research . Ann. Epidemiol. 26 , 301 – 305 .
Heintz-Buschart A. , Wilmes P. ( 2018 ). Human gut microbiome: Function matters . Trends Microbiol. 26 , 563 – 574 .
Hiippala K. , Jouhten H. , Ronkainen A. , Hartikainen A. , Kainulainen V. , Jalanka J. , Satokari R. ( 2018 ). The Potential of gut commensals in reinforcing intestinal barrier function and alleviating inflammation . Nutrients 10 , 988 .
Hildebrand F. , Nguyen T. L. , Brinkman B. , Yunta R. G. , Cauwe B. , Vandenabeele P. , Liston A. , Raes J. ( 2013 ). Inflammation-associated enterotypes, host genotype, cage and inter-individual effects drive gut microbiota variation in common laboratory mice . Genome Biol. 14 , R4 .
Hubbard T. D. , Murray I. A. , Perdew G. H. ( 2015 ). Indole and tryptophan metabolism: Endogenous and dietary routes to Ah receptor activation . Drug Metab. Dispos. 43 , 1522 – 1535 .
Hugenholtz F. , de Vos W. M. ( 2018 ). Mouse models for human intestinal microbiota research: A critical evaluation . Cell Mol. Life Sci. 75 , 149 – 160 .
Kilkenny C. , Browne W. J. , Cuthi I. , Emerson M. , Altman D. G. ( 2012 ). Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research . Vet. Clin. Pathol. 41 , 27 – 31 .
Kim S. , Goel R. , Kumar A. , Qi Y. , Lobaton G. , Hosaka K. , Mohammed M. , Handberg E. M. , Richards E. M. , Pepine C. J. , et al. . ( 2018 ). Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure . Clin. Sci. (Lond.) 132 , 701 – 718 .
Klaassen C. D. , Cui J. Y. ( 2015 ). Review: Mechanisms of how the intestinal microbiota alters the effects of drugs and bile acids . Drug Metab. Dispos. 43 , 1505 – 1521 .
Kundu P. , Blacher E. , Elinav E. , Pettersson S. ( 2017 ). Our gut microbiome: The evolving inner self . Cell 171 , 1481 – 1493 .
Li, F., Jiang, C., Krausz, K. W., Li, Y., Albert, I., Hao, H., Fabre, K. M., Mitchell, J. B., Patterson, A. D., and Gonzalez, F. J. (2013). Microbiome Remodelling Leads to Inhibition of Intestinal Farnesoid X Receptor Signalling and Decreased Obesity. Nat. Commun. 4 , 2384–2404.
Li C. Y. , Dempsey J. L. , Wang D. , Lee S. , Weigel K. M. , Fei Q. , Bhatt D. K. , Prasad B. , Raftery D. , Gu H. , et al. . ( 2018 ). PBDEs altered gut microbiome and bile acid homeostasis in male C57BL/6 mice . Drug Metab. Dispos. 46 , 1226 – 1240 .
Lindenbaum J. , Rund D. G. , Butler V. P. Jr , Tse-Eng D. , Saha J. R. ( 1981 ). Inactivation of digoxin by the gut flora: Reversal by antibiotic therapy . N. Engl. J. Med. 305 , 789 – 794 .
Lloyd-Price J. , Abu-Ali G. , Huttenhower C. ( 2016 ). The healthy human microbiome . Genome Med. 8 , 51 .
Lloyd-Price J. , Arze C. , Ananthakrishnan A. N. , Schirmer M. , Avila-Pacheco J. , Poon T. W. , Andrews E. , Ajami N. J. , Bonham K. S. , Brislawn C. J. , et al. .; IBDMDB Investigators. ( 2019 ). Multi-omics of the gut microbial ecosystem in inflammatory bowel disease . Nature 569 , 655 – 662 .
Lu K. , Abo R. P. , Schlieper K. A. , Graffam M. E. , Levine S. , Wishnok J. S. , Swenberg J. A. , Tannenbaum S. R. , Fox J. G. ( 2014 ). Arsenic exposure perturbs the gut microbiome and its metabolic profile in mice: An integrated metagenomics and metabolomics analysis . Environ. Health Perspect. 122 , 284 – 291 .
Melancon E. , Gomez De La Torre Canny S. , Sichel S. , Kelly M. , Wiles T. J. , Rawls J. F. , Eisen J. S. , Guillemin K. ( 2017 ). Best practices for germ-free derivation and gnotobiotic zebrafish husbandry . Methods Cell Biol. 138 , 61 – 100 .
Miyoshi J. , Bobe A. M. , Miyoshi S. , Huang Y. , Hubert N. , Delmont T. O. , Eren A. M. , Leone V. , Chang E. B. ( 2017 ). Peripartum antibiotics promote gut dysbiosis, loss of immune tolerance, and inflammatory bowel disease in genetically prone offspring . Cell Rep. 20 , 491 – 504 .
Miao J. , Ling A. V. , Manthena P. V. , Gearing M. E. , Graham M. J. , Crooke R. M. , Croce K. J. , Esquejo R. M. , Clish C. B. , Vicent D. , Biddinger S. B. ; Morbid Obesity Study Group. ( 2015 ). Flavin-containing monooxygenase 3 as a potential player in diabetes-associated atherosclerosis . Nat. Commun . 6 , 6498 .
Moya A. , Ferrer M. ( 2016 ). Functional redundancy-induced stability of gut microbiota subjected to disturbance . Trends Microbiol. 24 , 402 – 413 .
Mullane K. , Williams M. ( 2017 ). Enhancing reproducibility: Failures from reproducibility initiatives underline core challenges . Biochem. Pharmacol. 138 , 7 – 18 .
Nichols R. G. , Cai J. , Murray I. A. , Koo I. , Smith P. B. , Perdew G. H. , Patterson A. D. ( 2018 ). Structural and functional analysis of the gut microbiome for toxicologists . Curr. Protoc. Toxicol. 78 , e54 .
Norman J. M. , Handley S. A. , Baldridge M. T. , Droit L. , Liu C. Y. , Keller B. C. , Kambal A. , Monaco C. L. , Zhao G. , Fleshner P. , et al. . ( 2015 ). Disease-specific alterations in the enteric virome in inflammatory bowel disease . Cell 160 , 447 – 460 .
Ogilvie L. A. , Jones B. V. ( 2015 ). The human gut virome: A multifaceted majority . Front. Microbiol. 6 , 918 .
Orcutt R. , Gianni F. , Judge R. ( 1987 ). Development of an ‘Altered Schaedler Flora’ for NCI gnotobiotic rodents . Microecol. Ther. 17 , 59 .
Pascal V. , Pozuelo M. , Borruel N. , Casellas F. , Campos D. , Santiago A. , Martinez X. , Varela E. , Sarrabayrouse G. , Machiels K. , et al. . ( 2017 ). A microbial signature for Crohn’s disease . Gut 66 , 813 – 822 .
Peppercorn M. A. , Goldman P. ( 1972 ). The role of intestinal bacteria in the metabolism of salicylazosulfapyridine . J. Pharmacol. Exp. Ther. 181 , 555 – 562 .
Poesen R. , Claes K. , Evenepoel P. , de Loor H. , Augustijns P. , Kuypers D. , Meijers B. ( 2016 ). Microbiota-derived phenylacetylglutamine associates with overall mortality and cardiovascular disease in patients with CKD . J. Am. Soc. Nephrol. 27 , 3479 – 3487 .
Price N. D. , Magis A. T. , Earls J. C. , Glusman G. , Levy R. , Lausted C. , McDonald D. T. , Kusebauch U. , Moss C. L. , Zhou Y. , et al. . ( 2017 ). A wellness study of 108 individuals using personal, dense, dynamic data clouds . Nat. Biotechnol. 35 , 747 – 756 .
Proctor L. M. , Creasy H. H. , Fettweis J. M. , Lloyd-Price J. , Mahurkar A. , Zhou W. , Buck G. , Snyder M. P. , Strauss J. F. , Weinstock G. , et al. . ( 2019 ). The integrative human microbiome project . Nature 569 , 641 – 648 .
Qin J. , Li R. , Raes J. , Arumugam M. , Burgdorf K. S. , Manichanh C. , Nielsen T. , Pons N. , Levenez F. , Yamada T. , et al. .; MetaHIT Consortium. ( 2010 ). A human gut microbial gene catalogue established by metagenomic sequencing . Nature 464 , 59 – 65 .
Ridaura V. K. , Faith J. J. , Rey F. E. , Cheng J. , Duncan A. E. , Kau A. L. , Griffin N. W. , Lombard V. , Henrissat B. , Bain J. R. , et al. . ( 2013 ). Gut microbiota from twins discordant for obesity modulate metabolism in mice . Science 341 , 1241214 – 1241214 .
Robertson S. J. , Lemire P. , Maughan H. , Goethel A. , Turpin W. , Bedrani L. , Guttman D. S. , Croitoru K. , Girardin S. E. , Philpott D. J. ( 2019 ). Comparison of co-housing and littermate methods for microbiota standardization in mouse models . Cell Rep. 27 , 1910 – 1919 e2 .
Rogers G. B. ( 2015 ). The human microbiome: Opportunities and challenges for clinical care . Intern. Med. J. 45 , 889 – 898 .
Rosshart S. P. , Vassallo B. G. , Angeletti D. , Hutchinson D. S. , Morgan A. P. , Takeda K. , Hickman H. D. , McCulloch J. A. , Badger J. H. , Ajami N. J. , et al. . ( 2017 ). Wild mouse gut microbiota promotes host fitness and improves disease resistance . Cell 171 , 1015 – 1028 e13 .
Sanz Y. , Olivares M. , Moya-Perez A. , Agostoni C. ( 2015 ). Understanding the role of gut microbiome in metabolic disease risk . Pediatr. Res. 77 , 236 – 244 .
Schaffler H. , Breitruck A. ( 2018 ). Clostridium difficile - From colonization to infection . Front. Microbiol. 9 , 646 .
Schubert A. M. , Rogers M. A. , Ring C. , Mogle J. , Petrosino J. P. , Young V. B. , Aronoff D. M. , Schloss P. D. ( 2014 ). Microbiome data distinguish patients with Clostridium difficile infection and non- C. difficile -associated diarrhea from healthy controls . mBio 5 , e01021 – 14 .
Selwyn F. P. , Cui J. Y. , Klaassen C. D. ( 2015 ). RNA-seq quantification of hepatic drug processing genes in germ-free mice . Drug Metab. Dispos. 43 , 1572 – 1580 .
Sharpton S. R. , Ajmera V. , Loomba R. ( 2019 ). Emerging role of the gut microbiome in nonalcoholic fatty liver disease: From composition to function . Clin. Gastroenterol. Hepatol. 17 , 296 – 306 .
Sinha R. , Abu-Ali G. , Vogtmann E. , Fodor A. A. , Ren B. , Amir A. , Schwager E. , Crabtree J. , Ma S. , Abnet C. C. , et al. .; Microbiome Quality Control Project, Consortium. ( 2017 ). Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium . Nat. Biotechnol. 35 , 1077 – 1086 .
Sousa T. , Paterson R. , Moore V. , Carlsson A. , Abrahamsson B. , Basit A. W. ( 2008 ). The gastrointestinal microbiota as a site for the biotransformation of drugs . Int. J. Pharm. 363 , 1 – 25 .
Spanogiannopoulos P. , Bess E. N. , Carmody R. N. , Turnbaugh P. J. ( 2016 ). The microbial pharmacists within us: A metagenomic view of xenobiotic metabolism . Nat. Rev. Microbiol. 14 , 273 – 287 .
Stenman L. K. , Burcelin R. , Lahtinen S. ( 2016 ). Establishing a causal link between gut microbes, body weight gain and glucose metabolism in humans - Towards treatment with probiotics . Benef. Microbes 7 , 11 – 22 .
Stulberg E. , Fravel D. , Proctor L. M. , Murray D. M. , LoTempio J. , Chrisey L. , Garland J. , Goodwin K. , Graber J. , Harris M. C. , et al. . ( 2016 ). An assessment of US microbiome research . Nat. Microbiol. 1 , 15015 .
Tang W. H. , Wang Z. , Fan Y. , Levison B. , Hazen J. E. , Donahue L. M. , Wu Y. , Hazen S. L. ( 2014 ). Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-N-oxide in patients with heart failure: Refining the gut hypothesis . J. Am. Coll. Cardiol. 64 , 1908 – 1914 .
Tuddenham S. , Sears C. L. ( 2015 ). The intestinal microbiome and health . Curr. Opin. Infect. Dis. 28 , 464 – 470 .
Turner P. V. ( 2018 ). The role of the gut microbiota on animal model reproducibility . Animal Model Exp. Med. 1 , 109 – 115 .
Vandeputte D. , Kathagen G. , D’Hoe K. , Vieira-Silva S. , Valles-Colomer M. , Sabino J. , Wang J. , Tito R. Y. , De Commer L. , Darzi Y. , et al. . ( 2017 a). Quantitative microbiome profiling links gut community variation to microbial load . Nature 551 , 507 – 511 .
Vandeputte D. , Tito R. Y. , Vanleeuwen R. , Falony G. , Raes J. ( 2017 b). Practical considerations for large-scale gut microbiome studies . FEMS Microbiol. Rev. 41 , S154 – S167 .
Virgin H. W. ( 2014 ). The virome in mammalian physiology and disease . Cell 157 , 142 – 150 .
von Martels J. Z. H. , Sadaghian Sadabad M. , Bourgonje A. R. , Blokzijl T. , Dijkstra G. , Faber K. N. , Harmsen H. J. M. ( 2017 ). The role of gut microbiota in health and disease: In vitro modeling of host-microbe interactions at the aerobe-anaerobe interphase of the human gut . Anaerobe 44 , 3 – 12 .
Wallace B. D. , Wang H. , Lane K. T. , Scott J. E. , Orans J. , Koo J. S. , Venkatesh M. , Jobin C. , Yeh L. A. , Mani S. , et al. . ( 2010 ). Alleviating cancer drug toxicity by inhibiting a bacterial enzyme . Science 330 , 831 – 835 .
Wang Z. , Klipfell E. , Bennett B. J. , Koeth R. , Levison B. S. , DuGar B. , Feldstein A. E. , Britt E. B. , Fu X. , Chung Y.-M. , et al. . ( 2011 ). Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease . Nature 472 , 57 – 63 .
Wang X. , Yang Y. , Huycke M. M. ( 2017 ). Microbiome-driven carcinogenesis in colorectal cancer: Models and mechanisms . Free Radic. Biol. Med. 105 , 3 – 15 .
Ward T. L. , Dominguez-Bello M. G. , Heisel T. , Al-Ghalith G. , Knights D. , Gale C. A. ( 2018 ). Development of the human mycobiome over the first month of life and across body sites . mSystems 3 , e00140 – 17 .
Weitekamp C. A. , Phelps D. , Swank A. , McCord J. , Sobus J. R. , Catron T. , Keely S. , Brinkman N. , Zurlinden T. , Wheaton E. , et al. . ( 2019 ). Triclosan-resistant host-associated microbiota perform xenobiotic biotransformations in larval zebrafish . Toxicol. Sci . 172 , 109 – 122 .
Wilmanski T. , Rappaport N. , Earls J. C. , Magis A. T. , Manor O. , Lovejoy J. , Omenn G. S. , Hood L. , Gibbons S. M. , Price N. D. ( 2019 ). Blood metabolome predicts gut microbiome α-diversity in humans . Nat. Biotechnol . 37 , 1217 – 1228 .
Yang T. , Santisteban M. M. , Rodriguez V. , Li E. , Ahmari N. , Carvajal J. M. , Zadeh M. , Gong M. , Qi Y. , Zubcevic J. , et al. . ( 2015 ). Gut dysbiosis is linked to hypertension . Hypertension 65 , 1331 – 1340 .
Yassour M. , Vatanen T. , Siljander H. , Hamalainen A. M. , Harkonen T. , Ryhanen S. J. , Franzosa E. A. , Vlamakis H. , Huttenhower C. , Gevers D. , et al. .; on behalf of the DIABIMMUNE Study Group. ( 2016 ). Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability . Sci. Transl. Med. 8 , 343ra81 .
Yatsunenko T. , Rey F. E. , Manary M. J. , Trehan I. , Dominguez-Bello M. G. , Contreras M. , Magris M. , Hidalgo G. , Baldassano R. N. , Anokhin A. P. , et al. . ( 2012 ). Human gut microbiome viewed across age and geography . Nature 486 , 222 – 227 .
Zaneveld J. R. , McMinds R. , Vega Thurber R. ( 2017 ). Stress and stability applying the Anna Karenina principle to animal microbiomes . Nat. Microbiol . 2 , 1 – 8 .
Zhou W. , Sailani M. R. , Contrepois K. , Zhou Y. , Ahadi S. , Leopold S. R. , Zhang M. J. , Rao V. , Avina M. , Mishra T. , et al. . ( 2019 ). Longitudinal multi-omics of host microbe dynamics in prediabetes . Nature 569 , 663 – 671 .
Zhu A. , Sunagawa S. , Mende D. R. , Bork P. ( 2015 ). Inter-individual differences in the gene content of human gut bacterial species . Genome Biol. 16 , 82 .
Zimmermann M. , Zimmermann-Kogadeeva M. , Wegmann R. , Goodman A. L. ( 2019 ). Mapping human microbiome drug metabolism by gut bacteria and their genes . Nature 570 , 462 – 467 .
Author notes
- biological markers
- xenobiotics
- biotransformation
- intestinal bacteria
- toxic effect
Email alerts
Citing articles via.
- Recommend to your Library
Affiliations
- Online ISSN 1096-0929
- Print ISSN 1096-6080
- Copyright © 2024 Society of Toxicology
- About Oxford Academic
- Publish journals with us
- University press partners
- What we publish
- New features
- Open access
- Institutional account management
- Rights and permissions
- Get help with access
- Accessibility
- Advertising
- Media enquiries
- Oxford University Press
- Oxford Languages
- University of Oxford
Oxford University Press is a department of the University of Oxford. It furthers the University's objective of excellence in research, scholarship, and education by publishing worldwide
- Copyright © 2024 Oxford University Press
- Cookie settings
- Cookie policy
- Privacy policy
- Legal notice
This Feature Is Available To Subscribers Only
Sign In or Create an Account
This PDF is available to Subscribers Only
For full access to this pdf, sign in to an existing account, or purchase an annual subscription.
- Reference Manager
- Simple TEXT file
People also looked at
Systematic review article, recent advanced technologies for the characterization of xenobiotic-degrading microorganisms and microbial communities.

- 1 State Key Laboratory for Conservation and Utilization of Subtropical Agro-Bioresources, Guangdong Province Key Laboratory of Microbial Signals and Disease Control, Integrative Microbiology Research Centre, South China Agricultural University, Guangzhou, China
- 2 Guangdong Laboratory for Lingnan Modern Agriculture, Guangzhou, China
Global environmental contamination with a complex mixture of xenobiotics has become a major environmental issue worldwide. Many xenobiotic compounds severely impact the environment due to their high toxicity, prolonged persistence, and limited biodegradability. Microbial-assisted degradation of xenobiotic compounds is considered to be the most effective and beneficial approach. Microorganisms have remarkable catabolic potential, with genes, enzymes, and degradation pathways implicated in the process of biodegradation. A number of microbes, including Alcaligenes, Cellulosimicrobium, Microbacterium, Micrococcus, Methanospirillum, Aeromonas, Sphingobium, Flavobacterium, Rhodococcus, Aspergillus, Penecillium, Trichoderma, Streptomyces, Rhodotorula, Candida , and Aureobasidium , have been isolated and characterized, and have shown exceptional biodegradation potential for a variety of xenobiotic contaminants from soil/water environments. Microorganisms potentially utilize xenobiotic contaminants as carbon or nitrogen sources to sustain their growth and metabolic activities. Diverse microbial populations survive in harsh contaminated environments, exhibiting a significant biodegradation potential to degrade and transform pollutants. However, the study of such microbial populations requires a more advanced and multifaceted approach. Currently, multiple advanced approaches, including metagenomics, proteomics, transcriptomics, and metabolomics, are successfully employed for the characterization of pollutant-degrading microorganisms, their metabolic machinery, novel proteins, and catabolic genes involved in the degradation process. These technologies are highly sophisticated, and efficient for obtaining information about the genetic diversity and community structures of microorganisms. Advanced molecular technologies used for the characterization of complex microbial communities give an in-depth understanding of their structural and functional aspects, and help to resolve issues related to the biodegradation potential of microorganisms. This review article discusses the biodegradation potential of microorganisms and provides insights into recent advances and omics approaches employed for the specific characterization of xenobiotic-degrading microorganisms from contaminated environments.
Introduction
The environment is everything that naturally surrounds us and affects our daily lives on Earth. A safe and healthy environment is essential for the existence of life on this planet. However, in the era of advanced industrialization and urbanization, various anthropogenic activities are largely responsible for the introduction of toxic and hazardous pollutants such as environmental xenobiotics ( Embrandiri et al., 2016 ; Malla et al., 2018 ; Bhatt et al., 2020a ; Rodriguez et al., 2020 ). Xenobiotics are chemical substances not naturally produced or expected to be present within organisms. The term “xenobiotic” is usually used in the context of environmental pollutants to refer to synthetic compounds produced in large volumes for industrial, agricultural, and domestic use ( Embrandiri et al., 2016 ; Atashgahi et al., 2018 ; Dinka, 2018 ). There is growing public concern over the wide range of xenobiotic compounds being introduced, deliberately or accidentally, into the environment, which involves high potential risk to humans and animals ( Jacob and Cherian, 2013 ; Hashmi et al., 2017 ; Zhu et al., 2017 ; Dinka, 2018 ). Environmental xenobiotics include pesticides, polycyclic aromatic hydrocarbons (PAHs), pharmaceutical active compounds (PhACs), personal-care products (PCPs), phenolics, chlorinated compounds, and other industrial chemicals. Their increasing frequency in different environmental compartments has raised concerns about their potential adverse effects ( Crinnion, 2010 ; Kim et al., 2013 ; Embrandiri et al., 2016 ; Tsaboula et al., 2016 ; Dhakal et al., 2017 ). Their toxicity results in unprecedented health hazards and risks to environmental safety and security ( Godheja et al., 2016 ; Dovrak et al., 2017 ; Burgos-Aceves et al., 2018 ; Ravindra and Haq, 2019 ). Once xenobiotics are released into the environment, they can bioaccumulate within the food chain due to their high affinity toward organic substances, and produce toxic adverse effects toward natural ecosystems, humans, and animals ( Pedersen et al., 2003 ; Iovdijova and Bencko, 2010 ; Maurya, 2016 ). Consequently, they can cause severe chronic effects such as respiratory tract infections, damage to the immune system, pulmonary bronchitis, dysfunction of the nervous system, disruption of the endocrine system, behavioral and developmental disorders, and carcinogenic and mutagenic effects ( Sajid et al., 2015 ; Zhu et al., 2017 ; Dinka, 2018 ; Catron et al., 2019 ; Mishra et al., 2019 ; Bertotto et al., 2020 ). Thus, xenobiotic contamination represents a persistent anthropogenic threat and raises serious environmental concerns. Various physical and chemical treatment methods such as coagulation, filtration, adsorption, chemical precipitation, electrolysis ozonation, etc. have been used for the degradation and detoxification of such xenobiotic compounds, but not all these methods are very useful due to their high cost, waste-disposal problem and generation of toxic by-products that are sometimes more hazardous than the parent compound. In contrast, the biological remediation method, “bioremediation,” is a widely accepted clean-up strategy for the degradation of xenobiotics from contaminated environments without producing harmful products ( Paul et al., 2005 ; Perelo, 2010 ). Bioremediation involves the metabolic capabilities of microorganisms in the removal of pollutants and thus, is the most suitable and promising technology these days ( Gilliespie and Philp, 2013 ; Azubuike et al., 2016 ).
Microbial remediation of xenobiotic compounds is regarded as a superficial, proficient, economically feasible approach that uses a wide range of microorganisms to consume organic pollutants as carbon or nitrogen supplements to sustain their developmental activities ( Chen et al., 2013 ; Mahmoud, 2016 ; Arora et al., 2018 ; Ortiz-Hernandez et al., 2018 ; Siles and Margesin, 2018 ; Zhan et al., 2018 ; Bhatt et al., 2020b ). Microorganisms are ubiquitous in nature, and diverse microbial communities thrive in natural and extreme stress environments, including soil, water, the human gut, hydrothermal vents, acid mine runoff, and oil reservoirs ( Cycoń and Piotrowska-Seget, 2016 ; Jalowiecki et al., 2016 ; Ding et al., 2017 ; Aguinga et al., 2018 ; Wang Y. F. et al., 2018 ; Zierer et al., 2018 ; Delegan et al., 2019 ; Arora, 2020 ; Shekhar et al., 2020 ). Microbial populations exhibit potential for the remediation of any contaminated environment because of their genetic diversity and functionality ( Chen et al., 2015 ; Bastida et al., 2016 ; Bhatt and Barh, 2018 ; Dangi et al., 2018 ). Therefore, the study of microbial population existing in contaminated environments provides a significant knowledge of specific microbial characteristics that improve degradation rates. However, effective implementation of microbial remediation strategies needs advanced technical approaches, which provide an in-depth understanding about the dynamics aspects of microbial activity and survival under stressed environment ( Rastogi and Sani, 2011 ; Lima-Morales et al., 2016 ; Mao et al., 2019 ). The development in molecular, biotechnological, bioinformatics and system biology tools pertaining to bio-remedial problems have provided gene level mechanisms of bioremediation ( Ahmad and Ahmad, 2014 ; Aora and Bar, 2014 ; Singh D. P. et al., 2018 ; Jaiswal and Shukla, 2020 ; Nkongolo and Kotha, 2020 ; Wolf et al., 2020 ). Moreover, the direct study of microorganisms in a contaminated environment including the whole microbial population granted a new frontier of the scientific community to share the knowledge of the uncultured microbial world ( Zepeda et al., 2015 ; Zhao Q. et al., 2017 ; Panigrahi et al., 2019 ; Yan et al., 2020 ). The development of advance molecular tools and a better understanding of microbial metabolic and genetic structures and functions have accelerated encroachment in recombinant engineering techniques to enhanced bioremediation for removal of environmental pollutants ( Ram et al., 2005 ; Temperton and Giovannoni, 2012 ; Singh V. et al., 2018 ; Stein et al., 2018 ; Delegan et al., 2019 ; Marco and Abram, 2019 ; Puckett et al., 2020 ). Soil is the most dynamic environment for the enormous microbial population of immense diversity. It has been estimated that one gram of soil approximately contains 10 9 bacterial cells, but only <1% of these may be culturable in the laboratory ( Rossello-Mora and Amann, 2001 ). Culture based identification of diverse microbial population in a contaminated environment, is a challenging task, which is limited to fast-growing microbial diversity ( Gillbride et al., 2006 ). Thus, modern culture-independent molecular techniques represent a feasible approach to unrevealing the diversity and functional dynamics of microbial population in contaminated environments. Moreover, the advanced innovation in molecular tools and techniques provides new insights and changes the traditional research trend in the field of bioremediation ( Malik et al., 2008 ; Shah et al., 2011 ; Devarapalli and Kumavnath, 2015 ; Mahmoud, 2016 ; Biswas and Sarkar, 2018 ; Shakya et al., 2019 ). Omics technologies are the result of advanced molecular techniques, which involved direct characterization of genome structure of microorganisms, devoid of their culture sample ( Segata et al., 2013 ; Biswas and Sarkar, 2018 ; Jaiswal et al., 2019 ; Yu K. et al., 2019 ). Therefore, the applications of modern molecular techniques like metagenomic, transcriptomic, proteomic generates relevant information on genes and proteins expression levels in whole microbial communities under contaminated environments attempted to unravel the mechanism of microbial degradation and successful execution of bioremediation ( Keller and Hettich, 2009 ; Yang, 2013 ; Malla et al., 2018 ; Bharagava et al., 2019 ; Marco and Abram, 2019 ; Rodriguez et al., 2020 ). These methods are comparatively efficient, quicker, and accurate, which overcome the limitations of conventional molecular techniques. It explores the advanced microbial degradation mechanism of xenobiotics, their metabolic activities, genetic regulation and molecular-biology aspects ( Cycoń et al., 2017 ; Gutierrez et al., 2018 ; Gutleben et al., 2018 ; Mishra et al., 2020 ). Hence, this review article highlights the biodegradation potential of microorganisms and provides insights into recent advance methods of “omics” technologies employed in microbial degradation and remediation purpose of xenobiotics and their perspectives in modern biological research.
Bioremediation Potential of Microorganisms for Xenobiotic Compounds
The application of microorganisms in removing xenobiotics from soil, water or sediments through complete transformation or mineralization into harmless end products like CO 2 and H 2 O is a basic concept of bioremediation strategy ( Ortiz et al., 2013 ; Singh et al., 2016 ). Different microorganisms including bacteria ( Pseudomonas, Alcaligenes, Cellulosimicrobium, Microbacterium, Micrococcus, Methanospirillum, Aeromonas, Bacillus, Sphingobium, Flavobacterium , and Rhodococcus ), fungi ( Aspergillus, Penecillium, Trichoderma , and Fusarium ), and yeasts ( Pichia, Rhodotorula, Candida, Aureobasidium , and Exophiala ) have been reported to be involved in the efficient biodegradation of xenobiotic compounds from contaminated soil/water environments, due to their exceptional bioremediation potential ( Sathishkumar et al., 2008 ; Nzila, 2013 ; Sunita et al., 2013 ; Zhao Q. et al., 2017 ; Bharadwaj, 2018 ; Yang J. et al., 2018 ; Yang T. et al., 2018 ; Yu Y. et al., 2019 ; Bhatt et al., 2020c ). The biodegradation ability of microbes is greatly influenced by interactive ecological factors including soil, salinity, temperature, carbon source, moisture content, pH, nitrogen sources, inoculums concentration, etc. ( Megharaj and Naidu, 2010 ; Wu et al., 2014 ; Bhatt et al., 2019 ). Microorganisms harbor remarkable catabolic potential, genes, enzymes, and degradation pathways implicated in the process of bioremediation, which might be responsible in the evolution of novel traits and characters ( Widada et al., 2002 ; Scholer et al., 2017 ; Yan et al., 2018 ; Zhu et al., 2020 ). Moreover, microbial plasmids believed to be responsible for the continuous progression, evolution, and distribution of novel biodegradable genes/enzymes ( Zhang et al., 2016 ; Jeffries et al., 2019 ). These novel genes/enzymes have endowed microorganism's biodegradation capability to remove or detoxify a wide variety of environmental pollutants due to their inheritance horizontal gene transfer property ( Singh V. et al., 2018 ; Jaiswal et al., 2019 ; Li et al., 2019 ; Phale et al., 2019 ; French et al., 2020 ). The microbial remediation process can be further improved via successful application of genome editing and biochemical techniques that modify existing strains and result in the development of a genetically modified organism capable of simultaneously degrading several xenobiotics ( Shanker et al., 2011 ; Zhang et al., 2016 ; Hussain et al., 2018 ; Janssen and Stucki, 2020 ). The advancement of genetic manipulation technology gives more clear information and explores future prospects of bioremediation of xenobiotics through highly proficient microorganisms ( Sayler and Ripp, 2000 ; Shapiro et al., 2018 ; Wong, 2018 ; Liu et al., 2019 ). The chemical structures of several xenobiotic compounds are presented in Figure 1 .

Figure 1 . Chemical structures of several xenobiotic compounds.
Synthetic pesticides are the major example of xenobiotics especially the organochlorine pesticides (OCPs) that used extensively worldwide for a long period of time in agriculture as well as in insect control program. Several OCPs such as aldrin, dieldrin, dichloro diphenyl trichloro ethane (DDT), benzene hexachloride (BHC), and hexachlorocyclohexane are highly toxic in nature due to their stability and bioaccumulative property ( Aktar et al., 2009 ; Jayaraj et al., 2016 ; Awasthi and Awasthi, 2019 ; Pang et al., 2020 ). Lindane (γ-hexachlorocyclohexane) is one of the highly toxic organochlorine xenobiotic compound, which is well-studied for its microbial biodegradation through physical (soil structure, carbon and oxygen gradients, pH, and temperature) and chemical (dechlorination, dehydroxylation, and dehydrogenation) interactions ( Kaur and Kaur, 2016 ; Bashir et al., 2018 ; Zhang et al., 2021 ). The increasing concentration of lindane residues into the environment imposes severe health hazards such as carcinogenicity, mutagenicity, endocrine disruption, and immune-suppression diseases into the humans and other organisms ( Cuozzo et al., 2017 ; Zhang et al., 2020 ). Bacterial strains genera such as Bacillus, Burkholderia, Pseudomonas, Kocuria, Archromobacter, Sphingomonas, Chromohalobacter , demonstrated lindane biodegradation under axenic as well as anoxic conditions via dehydrogenation, dehydrochlorination, and hydroxylation, results in complete degradation or mineralization ( Giri et al., 2014 ; Cuozzo et al., 2017 ; Wang W. et al., 2018 ; Nagata et al., 2019 ; Zhang et al., 2020 ). A simplified catabolic pathway of lindane is presented in Figure 2 .

Figure 2 . Simplified catabolic degradation pathways of organochlorine and polycyclic aromatic hydrocarbon (PAH). Compounds (A) lindane (organochlorine) ( Endo et al., 2005 ; Geueke et al., 2013 ) and (B) naphthalene (PAH) ( Kiyohara et al., 1994 ; Dutta et al., 1998 ; Bamforth and Singleton, 2005 ; Baboshin et al., 2008 ).
Pyrethroids are broad spectrum pesticide mainly used against agricultural and household pests. Cypermethrin, cyhalothrin, deltamethrin, cyfluthrin, bifenthrin are the common example of synthetic pyrethroids ( Chen et al., 2012 , 2014 ; Bhatt et al., 2019 ; Zhan et al., 2020 ). These pesticides are highly toxic and persistent and can cause molecular toxicity, neurotoxicity, and reproductive toxicity ( Sharma et al., 2018 ; Bhatt et al., 2019 ; Gammon et al., 2019 ). One of the pyrethroid i.e., cypermethrin can cross the blood-brain barrier and induces neurotoxicity and motor deficits ( Singh et al., 2012 ). The persistence of these pesticides in the environment poses a severe threat to humans and other non-target terrestrial and aquatic organisms ( Burns and Pastoor, 2018 ; Ullah et al., 2018 ; Lu et al., 2019 ). Microbial strains such as Acinetobacter, Trichoderma, Roultella, Pseudomonas, Cunninghamella , and Bacillus have been reported for their efficient degradation of broad spectrum pesticides like cypermethrin and other pyrethroid pesticides through pyrethroid hydrolases ( Cycoń and Piotrowska-Seget, 2016 ; Zhan et al., 2018 ; Bhatt et al., 2019 ; Chen and Zhan, 2019 ). Co-metabolism exhibiting strains include Flavobacterium, Sphingomonas, Arthrobacter, Azotobacter, Achromobacter, Microbacterium, Brevibacterium, Rhodococcus, Trichoderma , and Aspergillus demonstrated their pollutant degradation capability, exclusive of pollutant consumption as an energy resource ( Nzila, 2013 ).
Polychlorinated bis-phenyles (PCBs) are classified as persistent organic pollutants with high toxicity ( Lallas, 2001 ; Pathiraja et al., 2019 ). They have been linked to chronic effects in humans including immune system damage, decreased pulmonary function, bronchitis, and interference with hormones leading to cancer ( Schecter et al., 2006 ). Microbial degradation of polychlorinated bis-phenyles (PCBs) is a promising remediation technology involving two major pathways: aerobic degradation and anaerobic dehalogination ( Abraham et al., 2002 ; Pathiraja et al., 2019 ). Bacterial strains Pseudomonas, Rhosocossus, Comamonas, Burkholderia , and Bacillus have been characterized for the oxidative degradation of PCBs through ring cleavage resulting into a common by product of chlorbenzoic acid ( Anyasi and Atagana, 2011 ; Jing et al., 2018 ).
Polycyclic aromatic hydrocarbons (PAHs) are potent environmental contaminants and xenobiotics that are widely distributed in the environment due to the incomplete combustion of organic matter. PAHs have moderate to high acute toxicity to aquatic life and birds ( Abdel-Shafy and Mansour, 2016 ; Pandey et al., 2017 ). Some PAHs such as Anthracene, benzo(a)pyrene, phenanthrene, and naphthalene are well-known to produce harmful biological effects such as genotoxicity, mutagenicity, and carcinogenicity and therefore pose a serious threat to the human health ( Kim et al., 2013 ; Lin et al., 2020 ). Microorganisms belonging to Sphingomonas, Sphingobium , and Novosphingobium have been found as efficient degrader of PAHs ( Lee et al., 2016b ; Fida et al., 2017 ; Auti et al., 2019 ). Rhodococcus, Cunninghamella, Pleurotus ostreatus, Oscillatoria, Agmenellum quadriplicatum, Brevibacterium , and Nocardiodes have been widely shown to metabolize particularly naphthalene and phenanthrene ( Ghosal et al., 2016 ; Siles and Margesin, 2018 ). Ortega-Gonzalez et al. (2015) demonstrated that Amycolaptosis sp. Poz14 degraded 100% of naphthalene and 37.87% of anthracene within 45 days. A PAH-degrading marine bacterium Cycloclasticus sp. has been isolated from sea sediments capable to breakdown xenobiotic naphthalene, pyrene, phenanthrene, and other aromatic hydrocarbons into their supplementary products through enzymatic pathways ( Wang W. et al., 2018 ). A simplified catabolic pathway of naphthalene is presented in Figure 2 .
Phthalates or esters of phthalic acids are synthetic xenobiotic chemicals, which are extensively used as plasticizers added to polyvinyl chloride to improve its flexibility and hardness ( Crinnion, 2010 ; Singh and Li, 2011 ; Przybylinska and Wyszkowski, 2016 ). Phthalates are readily released into the environment and create a risk of exposure for humans and other living organisms due their endocrine disrupting behavior ( Przybylinska and Wyszkowski, 2016 ). They cause infertility, reproductive and developmental toxicity in humans and animals ( Singh and Li, 2011 ; Przybylinska and Wyszkowski, 2016 ). Aerobic and anaerobic microbial degradation of xenobiotic phthalic acid, and isophthalaic acid considered as the most effective means of their removal from the environment ( Junghare et al., 2019 ; Boll et al., 2020 ). Di-2(ethyehxyl) phthalate (DEHP) is the most common member of phthalates, which are extensively used as plasticizer in plastics and disposable medical materials ( Singh and Li, 2011 ). DEHP is best known as an endocrine disrupter and can produce neural, heptotoxic, cardiotoxic, and carcinogenic effects on humans and animals ( Rowdhwal and Chen, 2018 ). Arthrobacter, Pseudomonas, Gordonia, Providencia, Acinetobacter, Microbacterium , and Rhodococuus have been identified as efficient DEHP degrading bacteria ( Nahurira et al., 2017 ; Yang T. et al., 2018 ).
In addition, the microbial consortium gained greater attention in bioremediation rather than pure microbial monocultures. The consortia cultures are better equipped in terms of metabolic and pollutant removal capability owing to their constant revelation of contaminant and promising mutual relationship with other available strains ( Patowary et al., 2016 ). Interestingly, microbial consortia can alleviate the metabolic limitations of single microbial culture and enhance biodegradation process by their miscellaneous assemblage of bacterial populations employed with extensive degradation potential ( Zafra et al., 2016 ; Li et al., 2020 ).
Recent Advanced Technologies Employed in Bioremediation for Identification and Characterization of Microorganisms and Microbial Communities
There are several revolutionary advanced molecular practices, including genomics, metagenomics, proteomics, transcriptomics, and metabolomics, which deliver deeper insights into microbial activities with respect to their genes, proteins, mRNA expression levels, enzymes and metabolic pathways with changing environments ( Figure 1 ). The integrated approach of these multiple technologies in the field of bioremediation is termed as the “omics approach,” used for the undeviating characterization of biological macromolecules, and their specific genetic and molecular structures and function mechanisms in a set of microorganisms/microbial communities ( Desai et al., 2009 ; Yang, 2013 ; Godheja et al., 2014 ; Franzosa et al., 2015 ; Chandran et al., 2020 ). The application of omics technologies provides comprehensive insights into microbial populations, their mechanisms of interaction with pollutants, metabolic activities, and genetic-regulation and molecular-biology aspects ( Akinsanya et al., 2015 ; Kaul et al., 2016 ; Misra et al., 2018 ; Marco and Abram, 2019 ; Huang et al., 2021 ). Moreover, these approaches can broaden our knowledge of the so-called “viable but non-culturable (VBNC)” bacteria and their potentially novel pathways for degrading environmental pollutants ( Oliver, 2010 ; Bodor et al., 2020 ). It is believed that these uncultured bacteria may play an important role in the biodegradation of environmental pollutant. However, little is known about the VBNC bacteria as these bacteria cannot be cultivated on conventional media and are very different from cells ( Su et al., 2013 ). VBNC cells exhibit metabolic and respiratory activities and may perform transcription and gene expression, which allows them to recover culturability ( Oliver et al., 2005 ; Zhao X. et al., 2017 ). The VBNC bacteria could be resuscitated in favorable conditions by an autoinducer (AI-2) ( Bari et al., 2013 ) or resuscitated promoting factor (Rpf) ( Li et al., 2015 ). The VBNC bacterial cells are detected on the basis of their viability. Several culture-independent molecular-based methods such as denaturing and temperature gradient gel electrophoresis (DGGE/TGGE), fluorescent in situ hybridization (FISH), terminal restriction fragment length polymorphism (T-RFLP), fatty acid methyl ester (FAME), and next-generation sequencing (NGS) technology are used for obtaining important information about the structural composition and genetic diversity of unculturable microorganisms ( Zhao X. et al., 2017 ; Bodor et al., 2020 ). Selective gene amplification is an emerging approach to detect viable cells. Zhong et al. (2016) developed a real-time fluorescence LAMP technique combined with PMA (propidium monoazide), a high-affinity photolysis DNA nucleic acid dye applied for the detection of VBNC V. parahaemolyticus . Thus, the combined use of these advanced molecular technologies with bioinformatic approaches increases understanding and brings in a new era of unrevealed soil microbial communities, as well as their associated mechanisms of biodegradation for their future applications in bioremediation ( Figure 3 ; Mocalli and Benedetti, 2010 ; Kumar et al., 2016 ; Dangi et al., 2018 ; Pandey et al., 2019 ; Pinu et al., 2019 ).

Figure 3 . Graphical presentation of integrated approach of advanced technologies in biodegradation of xenobiotic compounds.
Genomics and Metagenomics
Genomics and metagenomics are powerful tools for analyzing microbial communities at the genomic level from various contaminated environments. This technology gives a new array to environmental microbiologists for understanding unculturable microbiota with a genetic variability of microbial communities ( Devarapalli and Kumavnath, 2015 ; Zhu et al., 2018 ; Awasthi et al., 2020 ). It gives more details about the particular degradation potential of microbial communities, as it directly entails the whole-genome sequence from environmental samples ( Table 1 ). Metagenomic studies unblock traditional ways of uncultured microorganisms and explore their genetic advantage in the process of bioremediation ( Rahimi et al., 2018 ; Nascimento et al., 2020 ). Complete genome-sequence data of some important microbial strains, including Pseudomonas aeruginosa KT2440, Shewanella oneidensis MR-1, Deinococcus indicus R1, and Dehalococcoides mccartyi WBC-2 have already been given, which is pertinent to successful bioremediation ( http://www.tigr.org ). The new genes can tell too much about the degradation capability and substrate specificity.

Table 1 . Microorganisms and microbial communities using genomic and metagenomic approaches in biodegradation.
Current metagenomic practices allowed for identifying the whole-genome structure of microorganisms and specifying particular genes that are attributed to encode degradative enzymes for the mineralization of xenobiotics ( Zafra et al., 2016 ; Zhu et al., 2020 ). Thus, metagenomics clearly highlights the crucial role of novel genes in connecting the entire microbial population with functional diversity and structural identity. Metagenomics involves the manufacturing of metagenomic libraries that include (I) production of the proper size of DNA fragments, and ligation of these fragments into a suitable cloning vector; (II) further recombinant vectors introduced into an appropriate bacterium cloning host; (III) clones that harbor specific characters, functions, or sequences were screened for libraries ( Figure 3 ). Moreover, the screening of metagenomic libraries can be performed by two processes, i.e., sequence-driven analysis using high-throughput sequencing, and functional analysis using phenotypic expressions ( Handelsman, 2004 ). However, recent sequence-based metagenome analyses (such as SOLiD system of Applied Biosystems, Roche 454 sequencing) are performed without the construction of cloned libraries ( Kumar et al., 2020 ).
Function-driven metagenomics is a potent method for studying the functional aspect of genes. It is widely used for discovering novel genes with desired functions or exploring the sequence diversity of protein families ( Taupp et al., 2011 ; Lam et al., 2015 ). A function-driven analysis involves the construction and screening of metagenomic libraries to identify novel enzymes ( Chakraborthy and Das, 2016 ; Kumar et al., 2020 ). Using functional metagenomics, many novel antibiotic-resistant genes were identified from environmental sources ( Ngara and Zhang, 2018 ). The majority of metagenome-derived hydrolytic enzymes, mainly esterases and glycoside hydrolases, have been characterized biochemically and mainly originated from functional metagenomics ( Steele et al., 2009 ; Taupp et al., 2011 ). A novel functional screening method, a metagenome extract thin layer chromatography (META) system, was developed by Rabausch et al. (2013) for the rapid detection of glycosyltransferase (GT) and other flavonoid-modifying enzymes from metagenomic clone libraries. It involves the screening of 38,000 clones from two different metagenomic libraries and allowed for the identification of two novel UDP glycosyltransferase (UGT) genes. Bouhajja et al. (2017) utilized function-based screening of metagenomic libraries to explore the diversity of genes and microorganisms involved in the monooxygenase-mediated toluene degradation in a hydrocarbon-polluted sediment sample.
Metagenomic approaches offer broad and reliable microbial identification on the species and strain levels, but they are much costlier methods, and the most challenging part is data investigation, which requires all short DNA sequences to pair together to assemble the final genome structure ( Bragg and Tyson, 2014 ). The involvement of indigenous soil microorganisms in the degradation of PAHs was undertaken by Zafra et al. (2016) through a metagenomic approach. This study demonstrated the biodegradation efficiency of microbial consortia with their degradative enzymes and metabolites generated during the remediation process. The microbial-community dynamics of refined- and crude-petroleum-contaminated soil by next-generation sequencing (NGS), and their capability to degrade hydrocarbons and plant-growth-promotion potential through in-silico analysis was investigated by Auti et al. (2019) . In this study, 16S rRNA amplicon sequencing on the Illumina MiSeq platform and PICRUSt revealed that both types of soil contained microbial communities with excellent metabolic potential for petroleum hydrocarbon (PHC) degradation. Using KEGG orthology, the abundance of functional genes involved in hydrocarbon degradation showed the presence of 61 enzyme-encoding genes, such as alkane monooxygenase, alcohol dehydrogenase, and aldehyde dehydrogenase ( Auti et al., 2019 ). 16S rDNA or 16S rRNA gene sequencing has led to evolutionary insights into the phylogenetic and taxonomic identification of microorganisms. The 16S rRNA gene consists of several highly conserved regions interleaved with variable regions in all microorganisms and thus is highly suited as a target gene for sequencing DNA ( Fuks et al., 2018 ; Gursoy and Can, 2019 ). The bacterial 16S rRNA gene generally contains nine “hypervaiable regions” that demonstrate the considerable sequence diversity of bacterial species and can be used for species identification. The 16 S rRNA gene sequence similarity between two strains provides a simple yet robust criterion for the identification of newly isolated strains, whereas phylogenetic analyses can be used to elucidate the overall evolutionary relationship between related taxa ( Johnson et al., 2019 ). Thus, 16S rRNA gene sequencing analysis is a highly recommended cost-effective technique for the phylogenetic resolution and taxonomic profiling of microbial communities ( Auti et al., 2019 ).
The NGS approach has completely changed microbial-community analysis, as it provides comparative details in terms of temporal and spatial data ( Hidalgo et al., 2020 ). There are several NGS technologies, including Illumina, Ion Torrent, SOLiD, and 454 ( Caporaso et al., 2012 ; Liu et al., 2012 ; Knief, 2014 ; Salipante et al., 2014 ; Machado et al., 2019 ). These are high-throughput sequencing techniques of ribosomal genes that quantify community structures and functions at a higher resolution, e.g., 16S rRNA in prokaryotes, and 5S or 18S rRNA genes, or the internal-transcribe-spacer (ITS) region in eukaryotes ( Luo et al., 2012 ). The effectiveness of such NGS technologies in analyzing microbial communities from diverse environments was elucidated, validated, and documented in many studies ( Brown et al., 2013 ; Shokralla et al., 2014 ; Zhou et al., 2015 ; Niu et al., 2016 ; Scholer et al., 2017 ). In addition, PacBio (Pacific Biosciences) and Oxford Nanopore are highly advanced, reliable, and accurate third-generation sequencing platforms applied to microbial community analysis ( Lu et al., 2016 ; Chandran et al., 2020 ). Oxford Nanopore Technologies has launched a portable MinION USB nanopore that does not rely on DNA replication and has the advantage of reading full-length molecules in real time. PacBio RS II, the first commercialized genomic sequencer, developed by Pacific Biosciences, uses single-molecule, real-time (SMRT) technology and is able to sequence single DNA molecules in real time without PCR amplification ( Wagner et al., 2016 ; Nakano et al., 2017 ). The complete genome sequence of atrazine-degrading Arthrobacter sp. ZXY-2 ( Zhao X. et al., 2017 ) and organophosphate-degrading Sphingobium fuliginis ATCC 27552 ( Azam et al., 2019 ) was analyzed using the PacBio RSII sequencing platform to gain more insight into the genetic basis and unravel its degradation potential.
Jeffries et al. (2019) performed functional metagenomic studies in pesticide-contaminated soil to explore the degradation rates of organophosphorus xenobiotic compounds. Their study demonstrated that two distinct soil groups had different functional and taxonomic profiles, and predicted biodegradation potential in rapidly and slowly degrading soil clusters. Burkholderia, Acidomicrobium, Koribacter , and Bradyrhizobium were most abundantly present in rapidly degrading clusters, whereas Singulisphaera, Solibacter , and Desulfomonile were in slowly degrading clusters. The degradation assays of organophosphorus also suggested that slow-degradation clusters had significantly higher abundances of virulence genes and metabolic pathways for fatty acids and carbohydrates. In contrast, rapid-degradation clusters contain more abundant genes related to the transposable elements, membrane transport, and nutrient cycling of nitrogen and phosphorus enzymes potentially involved in phosphorus metabolism. Moreover, rapid-degradation soils also showed a higher abundance of genes encoding phosphodiesterase enzymes, which cleave phosphodiester bonds present in organophosphorus and play a major role in pesticide degradation. Overall, this study gives an overall framework of metagenomic approaches to predict the microbial degradation of xenobiotic organophosphorus compounds.
Metagenomic analysis of a complex community of lindane-contaminated pond sediment was conducted by Negi and Lal (2017) through comparative genomics. The results of this study revealed genomic variation present in pond sediment with degradative enzymes (hydrolases, isomerases, lyases, and oxidoreductases) involved in the biodegradation of hexachlorocyclohexane and chlorobenzene (ko00361), and other xenobiotic compounds. Cellulomonas, Micrococcus, Nocardioides, Kribbella, Isoptericola, Clavibacter, Gutenbergia, Streptomyces, Sanguibacter , and Kineococcus were found to be the most dominating genera present in the aromatic-hydrocarbon-contaminated pond sediment. Genes involved in lindane metabolization, enriched with sequences for linA and linB , were also found in the pond-sediment metagenome.
Whole-metagenome sequencing of e-waste-contaminated microbial populations was conducted by Salam and Verma (2019) , who demonstrated that the functional diversity and structural composition of microorganisms significantly changes due to the detrimental impact of e-waste. Denaturing gel gradient electrophoresis (DGGE) community-profiling results revealed that bacterial groups such as Proteobacteria, Firmicutes, Bacteroidetes , and Chloroflexi were decreased. Zhu et al. (2020) explored microbial assemblage and functional genes potentially involved in upstream and downstream phthalate degradation in soil via a metagenomic approach. Results indicated that bacterial taxon Actinobacteria ( Pimelobacter, Nocardioides, Gordonia, Nocardia, Rhodococcus , and Mycobacterium) was a major degrader under aerobic conditions, and bacterial taxa Proteobacteria ( Ramlibacter and Burkholderia ), Acidobacteria, and Bacteroidetes were involved under anaerobic conditions. The members of Geobacteraceae and Peptococcaceae microbiota present in the jet-fuel-contaminated site could be exploited for their remarkable metabolic potential for the mitigation of toluene and benzene, as exposed by metagenomic analysis ( Hidalgo et al., 2020 ).
Transcriptomics
Transcriptomics is a remarkable tool that addresses the division of genes transcribed in any organism known as the transcriptome. It provides functional insight links involving the genome, proteome, and cellular phenotype by studying their mRNA transcriptional profiles, directly extracted from individual microbes or microbial communities ( Singh and Nagaraj, 2006 ; McGrath et al., 2008 ; Bashiardes et al., 2016 ). Significant changes were seen in gene-expression level and their regulation in microbial communities under stressful environments for their survival. Thus, transcriptomics and metatranscriptomics provide deep analysis of a genome wide range of differently expressed genes, either of the individual cell or the entire microbial community at a specific time ( Table 2 ; Li et al., 2014 ; He et al., 2015 ; Shakya et al., 2019 ). RNA seq and DNA microarrays are significantly powerful technologies to determine the mRNA expression level of every gene ( Diaz, 2004 ). GeoChip uses key enzymes or genes to spot various microbe-mediated mechanisms for biogeochemical cycles, resistance mechanism for heavy metals, and degradation pathways of xenobiotics ( He et al., 2010 ; Xiong et al., 2010 ; Xie et al., 2011 ). Similarly, DNA- and RNA-SIP (Stable Isotope Probing) technologies are also valuable to uncover the microbial taxa and catabolic genes that are important for the bioremediation of polluted environments ( Lueders, 2015 ).

Table 2 . Microorganisms and microbial communities using transcriptomic and metatranscriptomic approaches in biodegradation.
Dual RNA-seq transcriptional profile is a better approach to understand the basic nature and mechanism of differently expressed genes in the host and symbiotic microbes at a time ( Kaul et al., 2016 ). RNA seq allows for the detection of more differently expressed genes than a microarray alone does. Thus, recent advancements and developments in microarrays, RNA seq technology, transcriptomics, and metatranscriptomics revealed unexpected microbial diversity in aquatic and terrestrial environments with their synergistic relationships with humans, animals, plants, and other microorganisms ( Perez-Losada et al., 2015 ; White et al., 2016 ; Moniruzzaman et al., 2017 ; Berg et al., 2018 ; Crump et al., 2018 ).
RNA seq technology is considered more efficient than traditional microarray platforms in gene expression profiling as it provides a wider quantitative range of expression level changes compared to microarrays ( Roh et al., 2010 ; Shakya et al., 2019 ). The microarray technique requires a lot of effort and money to prepare custom-made microarrays. Furthermore, the target genes to be analyzed are limited in number and cannot cover the whole set of genes in the community. In contrast, a number of kits are now commercially available to carry out RNA-seq analysis, whereby a whole set of genes in the community can be quantitatively analyzed. Therefore, many studies are now performed by RNA-seq technology.
Lima-Morales et al. (2016) investigated the microbial organization and catabolic gene diversity of three types of contaminated soil under continuous long-term pollutant stress with benzene and benzene/toluene/ethylene/xylene (BTEX) to identify shifts in community structure and the prevalence of key genes for catabolic pathways. Moreover, de novo transcriptome synthesis gives new insights into and reveals basic information about non-model species without a genome reference. Hydrocarbon-degrading bacterium Achromobacter sp. was isolated from seawater, and indicated that the upregulation of enzymes such as dehydrogenases and monooxygenases, and novel genes associated with fatty acid metabolism is responsible for its enormous capability for hydrocarbon degradation and survival ( Hong et al., 2016 ).
Metatranscriptomic analysis of the wheat rhizosphere identified dominant bacterial communities of diverse taxonomic phyla, including Acidobacteria, Cyanobacteria, Bacteroidetes, Steptophyta, Ascomycota, and Firmicutes, having functional roles in the degradation of various xenobiotic pollutants ( Singh D. P. et al., 2018 ). Multiple enzymes such as isomerases, oxygenases, decarboxylases, reductases, kinases, and inner membrane translocators were identified that were associated with 21 different pathways for benzoates, aromatic amines, phenols, bisphenols, and other xenobiotics ( Singh et al., 2016 ). An et al. (2020) elucidated the study of the transcriptome for the characterization of hexaconazole degrading strain Sphingobacterium multivorum , obtained from activated sludge. This strain was capable of degrading 85.6% hexaconazole in just 6 days and of generating three different metabolites, M1, M2, and M3, recognized as (2-(2,4-dichlorophenyl)-1-(1H-1,2,4-triazol-1-yl)hexane-2,5diol), (2-(2,4-dichlorophenyl) hexane-1,2-diol), and (1H-1,2,4-triazol), respectively. The results of transcriptome sequencing revealed the presence of 864 differential genes in which dehydrogenases, aldehydes, monooxygenases, and RND and AC transporters were upregulated. The M1 metabolite was perhaps generated due to the participation of monooxygenases.
Genomic and transcriptomic approaches were used by Sengupta et al. (2019) for gaining mechanistic insight into 4-nitrophenol (4-NP) degrading bacterium Rhodococcus sp. strain BUPNP1. This study identified a catabolic 43 gene cluster named nph that harbors not only mandatory genes for the breakdown of 4-NP into acetyl co-A and succinate by nitrocatechol, but also for other diverse aromatic compounds. An integrated approach of metagenomics and metatranscriptomics revealed the metabolic capabilities and synergistic relationship between Sphingomonas spp., Pusillimonas sp., and Pseudomonas sp. in the degradation of bisphenol A (BPA) ( Yu K. et al., 2019 ).
Metatranscriptomic analysis of this interaction model demonstrated genes encoding the transcription of 1,2-bis(4-hydroxyphenyl)-2-propanol (1-BP) into 4-hydroxybenzaldehyde (4-HBD) and 4-hydroxy-acetophenone (4-HAP) via 3,4-dihydroxybenzoate (3,4-DHB) and 3-oxoadipate (3-ODP), respectively, to the tricarboxylic acid cycle (TCA)cycle. Marcelino et al. (2019) identified fungal species and subspecies in a mixed community by using metatranscriptomics. This study suggested a strain-level discrepancy between the Cryptococcus fungal species and their in-situ mock communities. Thus, transcriptomic analysis provides a large amount of gene information about the potential function of microbial communities in adaptation and survival in extreme environments ( Singh D. P. et al., 2018 ; Mao et al., 2019 ; Marcelino et al., 2019 ).
Proteins are crucial effectors of biological responses, stabler than RNAs in living organisms, and possibly confer an integral view of biological function expressed in situ ; the term proteomics is put forward to study the entire set of proteins expressed in an organism ( Ram et al., 2005 ; Singh, 2006 ; Hettich et al., 2013 ). Thus, proteomics has emerged as an interesting and fruitful technology to study protein expression (post-translational modifications, protein turnover, proteolysis, and changes in the corresponding gene expression) of the microbial world ( Keller and Hettich, 2009 ; Aslam et al., 2017 ). Proteomics is a promising aspect of omic technologies in the field of microbiology, allowing for investigating the complete protein profile obtained in a straight line from a composite microbial population in a contaminated environment ( Williams et al., 2013 ; Arsene-Ploetze et al., 2015 ; Wang et al., 2016 ). However, metaproteomics is used to decipher crucial information regarding the protein profiling of two diverse ecological units ( Arsene-Ploetze et al., 2015 ). Proteomics has been used to identify microbial communities/microorganisms in various ecosystems including soil and sediment, activated sludge, marine and groundwater sediment, acid mine biofilms, and wastewater plants, as illustrated in Table 3 ( Williams et al., 2013 ; Colatriano et al., 2015 ; Grob et al., 2015 ; Bastida et al., 2016 ; Jagadeesh et al., 2017 ). These studies revealed secret information related to protein synthesis, gene-expression stability, mRNA turnover, and protein–protein interaction networks in microbial communities in stress environments ( Aslam et al., 2017 ).

Table 3 . Microorganisms or microbial communities using proteomic and metaproteomic approaches in biodegradation.
The inclusion of a proteomic approach helps to identify related enzymes and their metabolic pathways in the bioremediation of xenobiotics from various contaminated sites ( Kim et al., 2004 ; Liu et al., 2017 ; Wei et al., 2017 ). Basically, there are four primary steps that involve proteomic analysis: (1) preparation of a biological sample; (2) extraction and separation of proteins by using two-dimensional gel electrophoresis (2D-GE); (3) protein gel images are examined by means of image-analysis software such as ImageMaster 2D or PDQuest; and (4) proteins are identified by using mass spectroscopy (MS)/MALDI-TOF/MS or LC-MS ( Yates et al., 2009 ; Chakka et al., 2015 ; Velmurgan et al., 2017 ).
A combined protein profile of 20 PAH-induced proteins was studied by proteomics in Mycobacterium vanbaalenii PYR-1 grown in a PAH-supplemented culture medium ( Kim et al., 2004 ). PAH exposure of five different compounds, i.e., pyrene, pyerene-4,5-quinone, phenanthrene, anthracene, and fluoranthere causes variation in protein composition, showing upregulation of multiple proteins for PAH treatment compared to an uninduced control sample. Several PAH-induced proteins were identified by LC-MS, including a catalase-peroxidase, a putative monooxygenase, a dioxygenase, a small subunit of naphthalene-inducible dioxygenase, and aldehyde dehydrogenase. The metaproteomic approach was employed by Bastida et al. (2016) to illustrate changes in metabolic activities during compost-treated bioremediation with the help of differential protein expressions in hydrocarbon-polluted soil. Metaproteomic analysis indicated that Sphingomonadales and uncultured bacteria are responsible for the degradation of hydrocarbons in compost-treated soil due to the higher expression of catabolic enzymes such as 2-hydroxymuconic semialdehyde, cis -dihydrodiol dehydrogenase, and catechol 2,3-dioxygenase, dioxygenases involved in the first oxygenation step of aromatic rings. Moreover, biphenyl-2,3-diol 1,2-dioxygenase, estradiol dioxygenase, and naphthalene 1,2-dioxygenase were identified in compost-treated samples. By using metaproteomics, this study explored the functional and phylogenetic relationship of contaminated soil, and the microbial key players involved in compost-assisted bioremediation.
Another study, undertaken by Vandera et al. (2015) demonstrated the comparative proteomic analysis of Arthrobacter phenanivorans Sphe3 on aromatic compounds phenanthrene and phthalates. The proteomic approach confirmed the involvement of several proteins in aromatic-substrate degradation by identifying those mediating the initial ring hydroxylation and ring cleavage of phenanthrene to phthalate. This study also revealed the presence of both the ortho- and the meta- cleavage pathway for the degradation of these aromatic compounds, and it also identified all proteins that take part in these pathways and are highly upregulated upon phthalate growth in comparison to phenanthrene growth.
Proteomic analysis of pyrene-degrading bacterium Achromobacter xylosoxidans PY4 was performed by Nzila et al. (2018) , who identified a total of 1,094 proteins. Among these, 95 proteins were detected in glucose supplementation, and 612 proteins were detected in the presence of pyrene. Furthermore, 25 upregulated proteins were found to be involved in stress response and the progression of genetic information. Two upregulated proteins, 4-hydroxyphenylpyruvate dioxygenase and homogentisate 1,2-dioxygenase, are implicated in the lower degradation pathway of pyrene. Enzyme 4-hydroxyphenylpyruvate dioxygenase may catalyze the conversion of 2-hydroxybenzalpyruvic acid (metabolite of pyrene) to homogentisate. Homogentisate 1,2-dioxygenase is involved in the incorporation of 2 oxygen atoms to produce 4-maleyacetoacetate, which is an intermediate in several metabolic pathways.
Lee et al. (2016b) performed proteomic analysis of PAH-degrading bacterial isolate Sphingobium chungbukense DJ77. This strain exhibited outstanding degradation capability for various aromatic compounds. This study demonstrated the degradation of three xenobiotics compounds, i.e., phenanthrene, naphthalene, and biphenyls (PNB), and their associated proteins was analyzed by 2-DE and MALDI-TOF/MS analysis. During PNB biodegradation by bacterial cells, an alteration was observed in protein expression to cope with the stress condition. Comparative analysis of 2-DE results revealed that the intensity of 10 protein spots changes identically upon exposure to these xenobiotics in strain DJ77 ( Lee et al., 2016b ). Among these ten, five upregulated proteins with multiple functionalities were identified as putative dihydrodiol dehydrogenase (BphB), which catalyzes the NAD + -dependent oxidation of trans- dihydrodiols; 2,3-dihydrobisphenyl 1,2-dioxygenase (PhnQ), which cleaves the aromatic ring; and 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoate hydrolase (BphD), which degrades biphenyls and polychlorinated biphenyls. A part of the initial diverse catabolism of PNB by BphB, PhnQ, and BphD converged into the same catechol degradation branch. Now, catechol is first transformed into a ring-cleaved product, i.e., 2-hydroxymuconic semialdehyde by catechol 2,3-dioxygenase (PhnE). Moreover, it is assumed that this ring-cleaved product (2-hydroxymuconic semialdehyde) would be further degraded by 2-hydroxymuconic semialdehyde hydrolase (PhnD), and acetaldehyde dehydrogenase (PhnI) into a compound that can enter into the TCA cycle. Hence, these upregulated proteins, dehydrogenase, dioxygenase, and hydrolase, are involved in the catabolic degradation pathway of xenobiotics. The detection of intermediates from 2,3-dihydroxy-biphenyl degradation to pyruvate and acetyl-CoA by LC/MS analysis showed that ring-cleavage products entered the TCA cycle and were mineralized in strain DJ77. It was also suggested that strain DJ77 could completely degrade a wide range of PAHs via multiple catabolic pathways ( Lee et al., 2016b ).
The biodegradation mechanism of tetrabromobis-phenol A (TBBPA) was investigated in Phanerochaete chrysosporium by using a proteomic approach. iTRAQ quantitative analysis identified a total of 2,724 proteins in three biological samples. Compared to control TBBPA, stress caused 148 differentially expressed proteins in P. chrysosporium , among which 90 proteins were upregulated and 58 proteins were downregulated. The upregulation of cytochrome p450 monooxygenase, glutathione- S -transferase, O -methyltransferase, and other oxidoreductases is responsible for the biotransformation of TBBPA via oxidative hydroxylation and reductive debromination ( Chen et al., 2019 ).
A biodegradation study of endocrine-disrupting compound 4- n -nonylphenol (4- n - NP ) by filamentous fungus Metarhizium robertsii was investigated by Szewczyk et al. (2014) with proteomic analysis. This suggested that the main biodegradation mechanism involves the consecutive oxidation of the alkyl chain and benzene, which consequently results in the complete decomposition of the 4- n -NP compound. Proteomic profiling explored the involvement of nitro-reductase-like proteins related to the oxidation–reduction and ROS defense systems, and mainly engaged group of proteins in the removal of 4- n -NP. Proteomic data obtained in this study could not clearly explain the mechanism of 4- n -NP biodegradation in the tested fungal strain, but allowed for the formulation of hypotheses that the over-expressed enzymes in the cultures with 4- n -NP could play a role in xenobiotic removal and the biodegradation process.
Bioremediation of decabromodiphenyl ether (BDE-209) was explored in Microbacterium Y2 in a polluted water-sediment system through proteomics ( Yu Y. et al., 2019 ). Proteomic analysis showed that the overexpression of haloacid dehalogenases, glutathione s-transferase, and ATP-binding cassette (ABC) transporter might occupy important roles in BDE-209 biotransformation. Moreover, heat-shock proteins (HSPs), ribonuclease E, oligoribonuclease (Orn), and ribosomal proteins were activated to counter the BDE-209 toxicity. Thus, it is suggested that these proteins are implicated in microbial degradation, antioxidative stress, and glycolysis ( Yu Y. et al., 2019 ).
Metabolomics
Metabolomics is a well-established recent scientific technology, attributed toward the study of naturally occurring low-molecular weight (<1,000 Da) organic metabolites (organic acids; pyruvate, lactate, malate, formate, fatty acid-like acetate, etc.) inside a tissue, cell, or biofluid ( Johnson et al., 2011 ; Malla et al., 2018 ; Withers et al., 2020 ). Metabolomics explores the relationships between organisms and the environment, such as organismal responses to abiotic stressors, including both natural factors such as temperature, and anthropogenic factors such as pollution, to investigate biotic–biotic interactions such as infections, and metabolic responses ( Lindon et al., 2006 ; Griffiths, 2007 ; Mallick et al., 2019 ).
The combined use of metabolomics with these applications details the authentic collection of chemical outputs and inputs, which arbitrate the exchange of resources between the community and its host ( Table 4 ; Theriot et al., 2014 ; Wang Y. F. et al., 2018 ). Metabolomic studies in environmental sciences have been directed toward understanding changes in the concentration of metabolites associated with exposing model organisms to toxic compounds, such as xenobiotics ( Parisi et al., 2009 ; Seo et al., 2009 ).

Table 4 . Microorganisms or microbial communities using metabolomic approaches in biodegradation.
Metabolomics approach was utilized to investigate the degradation mechanism of carbaryl and other N -methyl carbamates pesticides in Burkholderia sp. strain C3 ( Seo et al., 2013 ). Metabolomes are dynamic and responsive to nutrient and environmental changes. The results of this study showed the metabolic adaptation of Burkholderia sp. C3 to carbaryl in comparison with glucose and nutrient broth. The metabolic changes were notably associated with the biosynthesis and metabolism of amino acids, sugars, PAH lipids, and cofactors. Differential metabolome analysis in response to different substrates identified 196 polar metabolites, 10 fatty acids, and 1 macromolecule (PHA) in this strain, and confirmed up-production metabolites in the pentose phosphate pathway, cysteine metabolism, amino acids, and disaccharides ( Seo et al., 2013 ). Thus, this metabolomic study could provide detailed insights into bacterial adaptation to different metabolic networks, and the metabolism of toxic pesticides and chemicals.
Environmental pollutants cause alterations in microbial communities, which consequently changes biochemical and metabolic functions in soil microorganisms. Microbial degradation of cyfluthrin by Photobacterium ganghwense was investigated via a comparative metabolic approach ( Wang et al., 2019 ). Metabolomics explored the biotransformation pathway of cyfluthrin with the identification of 156 metabolites during the biodegradation process. Recently, on the basis of interactions of indigenous soil microorganisms to PAH-contaminated soil, Li et al. (2018) elucidated that the majority of microbial metabolic functions were adversely affected to cope with PAH pollution. This study includes the combined study of enzyme activity and sequencing analysis with metabolomics, which further exposed the specific inhibition of soil metabolic pathways associated with carbohydrates, amino acids, and fatty acids due to microbial-community shifting under PAH stress.
Soil metabolomics is an effective approach to reveal the complex molecular networks and metabolic pathways operating in the soil microbial community. This approach can also be used to find biomarkers of soil contamination ( Jones et al., 2014 ). High-throughput sequencing and soil metabolomics investigated the differential structures and functions of soil bacterial communities in the pepper rhizosphere and bulk soil under plastic greenhouse vegetable cultivation (PGVC) ( Song et al., 2020 ). A total of 245 metabolites were identified, among which 11 differential metabolites were detected between rhizosphere and bulk soil, including organic acids and sugars that were positively and negatively correlated with the relative abundances of the differential bacteria. A starch and sucrose metabolic pathway was the most differentially expressed pathway in rhizospheric soil. The main functional genes participating in this pathway were predicted to be down regulated in rhizosphere soil. Sugar and organic acids as the main plant-root exudates in the rhizosphere, and they are also the main drivers of the shift in soil microbial community in the rhizosphere. These plant-root exudates act as an energy source to soil microbes, thus benefiting their growth ( Kuzyakov and Blagodatskaya, 2015 ; Shi et al., 2015 ). Linear discriminant analysis (LDA) effect size (LEfSe) analysis showed that bacterial phyla of Proteobacteria and Bacteroidetes were significantly higher in rhizosphere soil, benefitting plant growth. Thus, the relationship between soil metabolites and microbial communities guides the regulation of plant rhizoprocesses through soil amendments to increase plant growth.
Durand et al. (2010) conducted metabolic analysis of Bacillus sp. to characterize the metabolic pathway for the biodegradation of mesotrione, a herbicide. Analysis was carried out by using LC-NMR and LC-MS, and the result of these instrumental analyses was the identification of six metabolites, of which the structures of four metabolites were suggested. Szewczyk et al. (2017) performed metabolic analysis of fungal strain Metarhizium brunneum ARSEF 2017 to predict a biodegradation-pathway metabolic background for the removal of ametryn, an s -triazene herbicide. Qualitative LC-MS/MS metabolomic analysis of ametryn biodegradation resulted in the generation of four metabolites, i.e., 2-hydroxy atrazine, ethyl hydroxylated ametryn, S -demethylated ametryn, and diethyl ametryn.
Wright et al. (2020) evaluated the metabolomic characterization of two potent marine bacterial isolates, Mycobacterium sp. DBP42 and Halomonas sp. ATBC28, capable of the degradation of phthalate and plasticizers such as ATBC, DBP, and DEHP. That study presented the molecular analysis of metabolites generated during biodegradation. A metabolomic study confirmed that DBP and ATBC were degraded through the sequential removal of ester side chains, and generated monobutyl phthalate and phthalate in the case of DBP degradation, and citrate in the case of ATBC degradation in Mycobacterium sp. However, DEHP degradation did not follow the same pathway as that observed for DBP and ATBC. It was suggested that DEHP degradation is initiated through hydroxylation of the ester side chain by monooxygenase, and may occur via the β-oxidation of fatty acid side chains directly on the DEHP molecule. Moreover, in comparison with Mycobacterium sp., Halomonas sp. did not confirm any detectable degradation intermediates for the degradation of plasticizers and phthalate, but it harbored an array of enzymes suggested to be responsible for the degradation of other aromatic compounds. Therefore, metabolomic analyses demonstrated changes that occur in the composition of metabolites, aiding to fully understand the shifts mechanisms of metabolites during the microbial degradation or mineralization of environmental pollutants ( Lindon et al., 2006 ; Keum et al., 2008 ; d'Errico et al., 2020 ).
Bioinformatics
Bioinformatic technology developed a new array of computational technologies that uses both information technology and biological sciences. This modern technology seeks information from multiple high-throughput biological techniques, and keeps all biological data, helping to investigate and decide the relationship among organic molecules, including macromolecular sequences, biochemical and metabolic pathways, protein expressions, metabolites, and structures ( Fulekar and Geetha, 2008 ; Cooper et al., 2018 ; Dangi et al., 2018 ; Greene, 2018 ; Shekhar et al., 2020 ). Enormous amounts of data are generated from DNA, RNA, and protein sequences that need to be accurately executed; thus, bioinformatics has led to finding the best possible way to analyze such huge amounts of biological data via specific computational tools ( Aora and Bar, 2014 ; Bhatt et al., 2021 ). Therefore, bioinformatic-associated tools are very important to understand the bioremediation of toxic pollutants. Bioinformatics provides superior information regarding the cellular, molecular, and genetical bases of xenobiotic degradation and detoxification ( Kumar et al., 2016 ; Huang et al., 2020 ). There are a number of bioinformatic tools and applications that are available to use for biodegradation studies, as listed in Table 5 .

Table 5 . Bioinformatic databases and software tools used in biodegradation studies.
MetaRouter is one of such application that is freely open and a modular architecture to a variety of consumers from any place in a safe and secure manner, just by connecting to an Internet server ( Pazos et al., 2005 ). For the analysis of biodegradation studies, many bioinformatic resources are exclusively available. The University of Minnesota Biocatalysts/Biodegradation Database (UM-BBD) was introduced in 1995 and contains information regarding microbial catabolism and related biotransformation, biodegradation reactions, catabolic enzymes, and pathways for xenobiotics and other hazardous pollutants of various microorganisms. This database is connected to several other databases, such as BRENDA, ENZYME, ExPASy, and NCBI, to collect and store information related to gene structure and enzymes that take part in the biodegradation of environmental contaminants ( Ellis et al., 2006 ). Genomic sequences of microorganisms with competent and efficient degradation abilities could be easily investigated via another widely used database, the National Center for Biotechnology Information (NCBI). It gives a detailed and complete pipeline for annotations, and comprehensive analysis of more than 6,000 microbial genomes ( Brown et al., 2015 ). PRIDE ( Vizcaino et al., 2016 ) is the world's largest data repository of mass-spectroscopy-based proteomic data, and MetaboLights ( Kale et al., 2016 ) is a database for metabolomic experiments and derived information. The GenBank database is freely available in NCBI, and it provides most up-to-date and comprehensive DNA sequence information ( Benson et al., 2013 ). Recently, there have been a number of databases such as CAMERA, MG-RAST, and IMG/M that were developed and employed for the analysis and in-depth understanding of diverse microbial populations, metabolic reconstruction, taxonomic affiliations, and their inter- and intra-relationship networks. Another database, Bionemo, developed by the structural computational-biology group at the Spanish National Cancer Research Center, gives information related to specific genes and proteins that take part in biodegradation reactions and metabolic pathways ( Carbajosa et al., 2009 ). It provides insights into sequences, domains, protein structures, and regulatory elements, and transcription factors for their respective genes. Integrated bioinformatic approaches are employed for the metagenomic characterization of the soil microbial communities of different soil sites by using MetaPhlAn, KEGG, XLSX, and LEfSe bioinformatic databases to reveal the ancestral and functional characterization of diverse soil microbial populations ( Arora et al., 2009 ; Xu et al., 2014 ; Kumar et al., 2016 ). The degradation or detoxification of xenobiotic pollutants through microbial communities is a highly considered and proficient remediation technology, and there is no single resource accessible that provides all the information with reference to environmental contaminants, microorganisms, and their bioremediation potentialities. Thus, these databases coalescing the detailed information about the nature of pollutants, their metabolic pathways, bioremediation microorganisms, catabolic genes, enzymes, and protein-expression profiles would be a significant tool to open up a new vista and enlighten future research science in the field of bioremediation.
Conclusions
Microbial communities have great potential to mediate the successful biodegradation process of xenobiotic-contaminated soil/water environments. However, the greater part of mainstream microorganisms involved in bioremediation are still undefined because not all organisms in nature could be cultured under in vitro environments, but reside in viable-but-non-culturable (VBNC) environments. Thus, to explore the hidden knowledge of these VBNC organisms, recent advanced practices and sophisticated up-to-date technologies are highly desirable to understand the genetic and molecular biology of microorganisms. Newly developed molecular techniques offer promising approaches to address the in-depth characterization of microbial communities from molecule to gene. Recent omics technologies such as metagenomics, transcriptomics, and proteomics are helpful in obtaining information about nucleic acids, enzymes, catabolic genes, plasmids, and metabolic machineries and metabolites generated during the biodegradation process. However, the solitary employment of any individual omics technology is not sufficient to explore or illustrate secret information regarding microbial-remediation practices. Therefore, an interdisciplinary application of multiple omics studies highlights the perspectives of system biology for providing an integrative understanding between genes, proteins, and environmental factors responsible for the whole microbial-degradation process, and gives a new array of novel technologies, such as genome-editing and next-generation-sequencing tools CRISPR-Cas9, TALEN, and ZFNs, which are potent gene-editing tools that design microbes with specific degradation-function genes and provide unique insights into microbial remediation. Moreover, the successful execution of omics technologies could not be possible without the use of bioinformatic tools. The establishment of informative genomic and proteomic databases has been revolutionized by bioinformatics, which facilitates broad information about cellular- and metabolic-mechanism pathways for environmental pollutants. Hence, the involvement of these advanced technologies in the biological sciences shows the way to next-level research in the bioremediation potential of microorganisms, and exploits their capability to remove xenobiotic contamination.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Author Contributions
Conceptualization and writing—original draft preparation: SM. Writing—review and editing: ZL, SP, WZ, PB, and SC. Supervision, funding acquisition, and project administration: SC. All authors contributed to the article and approved the submitted version.
This study was funded by the Key-Area Research and Development Program of Guangdong Province, China (2018B020206001), the National Natural Science Foundation of China (31401763), and the Guangdong Special Branch Plan for Young Talent with Scientific and Technological Innovation, China (2017TQ04N026).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abdel-Shafy, H., and Mansour, M. S. M. (2016). A review on polycyclic aromatic hydrocarbons: source, environmental impact, effect on human health and remediation. Egyp. J. Petroleum 25, 107–123. doi: 10.1016/j.ejpe.2015.03.011
CrossRef Full Text | Google Scholar
Abraham, W. R., Nogales, B., Golyshin, P. N., Piper, D. H., and Timmis, K. N. (2002). Polychlorinated biphenyl-degrading microbial communities in soil and sediments. Curr. Opin. Microbiol. 5, 246–253. doi: 10.1016/S1369-5274(02)00323-5
PubMed Abstract | CrossRef Full Text | Google Scholar
Aguinga, O. E., McMahon, A., White, K. N., Dean, A. P., and Pittman, J. K. (2018). Microbial community shifts in response to acid mine drainage pollution within a natural wetland ecosystem. Front. Microbiol. 9:1445. doi: 10.3389/fmicb.2018.01445
Ahmad, A., and Ahmad, I. (2014). “Recent advances in the field of bioremediation”, in Biotechnology Vol. 11: Biodegradation and Bioremediation , ed M. Ahmad (India: Stadium Press LLC), 1–42.
Google Scholar
Akinsanya, M. A., Goh, J. K., Lim, S. P., and Ting, A. Y. (2015). Metagenomic study of endophytic bacteria in Aloe vera using next-generation technology. Genom. Data 6, 159–163. doi: 10.1016/j.gdata.2015.09.004
Aktar, W., Sengupta, D., and Chowdhary, A. (2009). Impact of pesticides use in agriculture: their benefits and hazards. Interdiscip. Toxicol. 2, 1–12. doi: 10.2478/v10102-009-0001-7
An, X., Tian, C., Xu, J., Dong, F., Liu, X., Wu, X., et al. (2020). Characterization of hexaconazole-degrading strain Sphingobacterium multivorum and analysis of transcriptome for biodegradation mechanism. Sci. Total Environ. 722:37171. doi: 10.1016/j.scitotenv.2020.137171
Anyasi, R., and Atagana, H. (2011). Biological remediation of polychlorinated bisphenyls (PCB) in the environment by microorganisms and plants. Afr. J. Biotechnol. 10, 18916–18938. doi: 10.5897/AJB10.557
Aora, P. K., and Bar, H. (2014). Integration of bioinformatics to biodegradation. Biol. Procedure Online 16:8. doi: 10.1186/1480-9222-16-8
Arkin, A. P., Stevens, R. L., and Cottingham, R. W. (2016). The DOE system biology knowledgebase (KBase). BioRxiv 12, 1–21. doi: 10.1101/096354
CrossRef Full Text
Arora, P. K. (2020). Bacilli-mediated degradation of xenobiotic compounds and heavy metals. Front. Bioeng. Biotech. 8:570307. doi: 10.3389/fbioe.2020.570307
Arora, P. K., Kumar, M., Chauhan, A., Raghava, G. P., and Jain, R. K. (2009). OxDBase: a database of oxygenases involved in biodegradation. BMC Res. Notes 2:67. doi: 10.1186/1756-0500-2-67
Arora, P. K., Srivastava, A., Garg, S. K., and Singh, V. P. (2018). Recent advances in degradation of chloronitrophenols. Bioresour. Technol . 250, 902–909. doi: 10.1016/j.biortech.2017.12.007
Arsene-Ploetze, F., Bertin, P. N., and Carapito, C. (2015). Proteomic tools to decipher microbial community structure and functioning. Environ. Sci. Pollut. Res. 22, 13599–13612. doi: 10.1007/s11356-014-3898-0
Aslam, B., Basit, M., Nisar, M. A., Khurshid, M., and Rasool, M. H. (2017). Proteomics: technologies and their applications. J. Chromatogr. Sci. 55, 182–196. doi: 10.1093/chromsci/bmw167
Atashgahi, S., Shetty, S. A., Hauke, S., and de Vos, W. M. (2018). Flux, impact and fate of halogenated xenobiotic compounds in the Gut. Front. Physiol. 9:888. doi: 10.3389/fphys.2018.00888
Auti, A. M., Narwade, N. P., Deshpande, N. M., and Dhotre, D. P. (2019). Microbiome and imputed metagenome study of crude and refined petroleum-oil contaminated soils: potential for hydrocarbon degradation and plant-growth promotion. J. Biosci. 44:114. doi: 10.1007/s12038-019-9936-9
Awasthi, K. R., and Awasthi, M. S. (2019). Health and environmental effects of organochlorine pesticides in developing countries. Open Access J. Environ. Soil Sci. 2, 206–208. doi: 10.32474/OAJESS.2019.02.000135
Awasthi, M. K., Ravindran, B., Sarsaiya, S., Chen, H., Wainaina, S., Singh, E., et al. (2020). Metagenomics for taxonomy profiling: tools and approaches. Bioengineered 11, 356–374. doi: 10.1080/21655979.2020.1736238
PubMed Abstract | CrossRef Full Text
Azam, S., Parthasarthy, S., Singh, C., Kumar, S., and Siddavattam, D. (2019). Genome organization and adaptive potential of archetypal organophosphate degrading Sphingobium fuliginis ATCC 27551. Genome Biol. Evol . 11, 2557–2562. doi: 10.1093/gbe/evz189
Azubuike, C. C., Chikere, C. B., and Okpokwasili, G. C. (2016). Bioremediation techniques- classification based on site of application: principle, advantages, limitations, and prospects. World. J. Microbiol. Biotechnol. 32, 180–180. doi: 10.1007/s11274-016-2137-x
Baboshin, M., Akimov, V., Baskunov, B., Born, T. L., Khan, S. U., and Golovleva, L. (2008). Conversion of polycyclic aromatic hydrocarbons by Sphingomonas sp. VKM B-2434. Biodegradation 19, 567–576. doi: 10.1007/s10532-007-9162-2
Bamforth, S. M., and Singleton, I. (2005). Bioremediation of polycyclic aromatic hydrocarbons: current knowledge and future directions. J. Chemical Technol. Biotechnol. 80, 723–736. doi: 10.1002/jctb.1276
Bari, S. M. N., Roky, M. K., Mohiuddin, M., Kamruzzaman, M., Mekalanos, J. J., and Faruque, S. M. (2013). Quorum-sensing autoinducers resuscitate dormant Vibrio cholera in environmental water samples. Proc. Natl. Acad. Sci. U.S.A. 110, 9926–9931. doi: 10.1073/pnas.1307697110
Bashiardes, S., Zilberman-Schapira, G., and Elinav, E. (2016). Use of metatranscriptomics in microbe research. Bioinf. Biol. Insights. 10, 19–25. doi: 10.4137/BBI.S34610
Bashir, S., Kuntze, K., Vogt, C., and Nijenhuis, I. (2018). Anaerobic biotransformation of hexachlorocyclohexane isomers by Dehalococcoides species and an enrichment culture. Biodegradation 29, 409–418. doi: 10.1007/s10532-018-9838-9
Bastida, F., Jehmlich, N., Lima, K., Moris, B. E., Richnow, H. H., Hernandez, T., et al. (2016). The ecological and physiological responses of the microbial community from a semiarid soil to hydrocarbon contamination and its bioremediation using compost amendment. J. Proteomic. 135, 162–169. doi: 10.1016/j.jprot.2015.07.023
Benson, D. A., Cavanaugh, M., Clark, K., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., et al. (2013). GenBank. Nucleic Acids Res. 41, D32–D37. doi: 10.1093/nar/gkt1030
Berg, C., Dupont, C. L., Asplund-Samuelsson, J., Celepli, N. A., Eiler, A., and Allen, A. E. (2018). Dissection of microbial community functions during a cyanobacterial bloom in the Baltic sea via metatranscriptomic. Front. Mar. Sci. 5:55. doi: 10.3389/fmars.2018.00055
Bertotto, L. B., Catron, T. R., and Tal, T. (2020). Exploring interactions between xenobiotics, microbiota and neurotoxicity in zebrafish. Neuro. Toxicol . 76, 235–244. doi: 10.1016/j.neuro.2019.11.008
Bharadwaj, A. (2018). “Bioremediation of xenobiotics: an eco-friendly cleanup approach”, in Green Chemistry in Environmental Sustainability and Chemical Education , eds V. S. Parmar, P. Malhotra, and D. Mathur (Singapore: Springer Nature), 1–13. doi: 10.1007/978-981-10-8390-7_1
Bharagava, R. N., Purchase, D., Saxena, G., and Mulla, S. I. (2019). “Application of metagenomic in microbial bioremediation of pollutants: from genomic to environmental cleanup”, in Microbial Diversity in the Genomic Era , eds S. Das and H. Dask (Elsevier; Academia Press), 459–477. doi: 10.1016/B978-0-12-814849-5.00026-5
Bhatt, P., and Barh, A. (2018). “Bioinformatic tools to study the soil microorganism: an in silico approach for sustainable agriculture”, in In-silico Approach for Sustainable Agriculture , eds D. K. Choudhary, M. Kumar, R. Prasad, and V. Kumar (Singapore: Springer), 169–182. doi: 10.1007/978-981-13-0347-0_10
Bhatt, P., Gangola, S., Bhandari, G., Zhang, W., Maithani, D., Mishra, S., et al. (2020a). New insights into the degradation of synthetic pollutants in contaminated environments. Chemosphere 259:128827. doi: 10.1016/j.chemosphere.2020.128827
Bhatt, P., Huang, Y., Hui, Z., and Chen, S. (2019). Insights into microbial applications for the biodegradation of pyrethroid insecticides. Front. Microbiol. 10:177. doi: 10.3389/fmicb.2019.01778
Bhatt, P., Joshi, T., Bhatt, K., Zhang, W., Huang, Y., and Chen, S. (2020b). Binding interaction of glyphosate with glyphosate oxidoreductase and C–P lyase: molecular docking and molecular dynamics simulation studies. J. Hazard. Mater. doi: 10.1016/j.jhazmat.2020.124927
Bhatt, P., Rene, E. R., Kumar, A. J., Zhang, W., and Chen, S. (2020c). Binding interaction of allethrin with esterase: bioremediation potential and mechanism. Bioresour. Technol. 315:123845. doi: 10.1016/j.biortech.2020.123845
Bhatt, P., Zhao, X., Huang, Y., Zhang, W., and Chen, S. (2021). Characterization of the role of esterases in the biodegradation of organophosphate, carbamate and pyrethroid pesticide. J. Hazard. Mater. 411:125026. doi: 10.1016/j.jhazmat.202.125026
Biswas, R., and Sarkar, A. (2018). “Omics' tools in soil microbiology-the state of art”, in Advances in Soil Microbiology: Recent Trends and Future Prospect, Microorganism for Sustainability , ed T. K. Adhya (Cham: Springer Nature), 35–64. doi: 10.1007/978-981-10-6178-3_3
Bodor, A., Bounedjoum, N., Vincze, E. G., Kis, A. E., Laczi, K., Bende, G., et al. (2020). Challenges of unculturable bacteria: environmental perspectives. Rev. Environ. Sci. Biotechnol. 19, 1–22. doi: 10.1007/s11157-020-09522-4
Boll, M., Geiger, R., Jughare, M., and Schink, B. (2020). Microbial degradation of phthalates: biochemistry and environmental implications. Environ. Microbiol. Rep. 12, 3–15. doi: 10.1111/1758-2229.12787
Bouhajja, E., McGuire, M., Liles, M. R., Bataille, G., Agathos, S. N., and George, I. F. (2017). Identification of novel toluene monooxygenase genes in a hydrocarbon-polluted sediments using sequence- and function-based screening of metagenomic libraries. Appl. Microbiol. Biotechnol. 101, 797–808. doi: 10.1007/s00253-016-7934-5
Bragg, L., and Tyson, G. (2014). “Metagenomics using next-generation sequencing,” in Environmental Microbiology Methods and Protocols , eds I. T. Paulsen and A. J. Holmes (Totowa, NJ: Humana Press), 183–201. doi: 10.1007/978-1-62703-712-9_15
Brown, G. R., Hem, V., and Katz, K. S. (2015). Gene: a gene-centered information resources at NCBI. Nucleic Acids Res. 43, D36–D42. doi: 10.1093/nar/gku1055
Brown, S. P., Callaham, M. A., Oliver, A. K., and Jumpponen, A. (2013). Deep ion torrent sequencing identifies soil fungal community shifts after frequent prescribed fires in a southeastern US forest ecosystem. FEMS Microbiol. Ecol. 86, 557–566. doi: 10.1111/1574-6941.12181
Burgos-Aceves, M. A., Cohen, A., Smith, Y., and Faggio, C. (2018). MicroRNAs and their role on fish oxidative stress during xenobiotic environmental exposure. Ecotoxicol. Environ. Saf. 148, 995–1000. doi: 10.1016/j.ecoenv.2017.12.001
Burns, C. J., and Pastoor, T. P. (2018). Pyrethroid epidemiology: a quality based review. Crit. Rev. Toxicol. 48, 297–311. doi: 10.1080/10408444.2017.1423463
Caporaso, J. G., Lauber, C. L., and Walters, W. A. (2012). Ultra-high-throughtput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J . 6, 1621–1624. doi: 10.1038/ismej.2012.8
Capsi, R., Billington, R., and Ferrer, L. (2016). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathways/genome databases. Nucleic Acids Res. 44, D471–D480. doi: 10.1093/nar/gkv1164
Carbajosa, G., Trigo, A., Valencia, A., and Cases, I. (2009). Bionemo: molecular information on biodegradation on the metabolism. Nucleic Acids Res. 37, D598–D602. doi: 10.1093/nar/gkn864
Castrejon-Godinez, M. L., Ortiz-Hernandez, M. L., Salazar, E., Encaracion, S., Mussali-Galante, P., Tovar-Sanchez, E., et al. (2019). Transcriptional analysis reveals the metabolic state of Burkholderia zhejiangensis CEIB S4-3 during methyl parathion degradation. Peer J. 7:e6822. doi: 10.7717/peerj.6822
Catron, T. R., Gaballah, S., and Tal, T. (2019). Using Zebra fish to investigate interactions between xenobiotics and microbiota. Curr. Pharmacol. Rep. 5, 468–480. doi: 10.1007/s40495-019-00203-7
Chakka, D., Gudla, R., Madikonda, A. K., Pandeeti, E. V. P., Partasarathy, S., Nandavaram, A., et al. (2015). The organophosphate degradation (opd) island-born esterase-induced metabolic diversion in Escherichia coli and its influence on p -nitrophenol degradation. J. Biol. Chem. 290, 29920–29930. doi: 10.1074/jbc.M115.661249
Chakraborthy, J., and Das, S. (2016). Molecular perspectives and recent advances in microbial remediation of persistent organic pollutants. Environ. Sci. Pollut. Res. 23, 16883–16903. doi: 10.1007/s11356-016-6887-7
Chandran, H., Meena, M., and Sharma, K. (2020). Microbial biodiversity and bioremediation assessment through omics approaches. Front. Environ. Chem . 1:570326. doi: 10.3389/fenvc.2020.570326
Chen, S., Chang, C., Deng, Y., An, S., Dong, Y. H., Zhou, J., et al. (2014). Fenpropathrin biodegradation pathway in Bacillus sp. DG-02 and its potential for bioremediation of pyrethroid-contaminated soils. J. Agric. Food Chem . 62, 2147–2157. doi: 10.1021/jf404908j
Chen, S., Deng, Y., Chang, C., Lee, J., Cheng, Y., Cui, Z., et al. (2015). Pathway and kinetics of cyhalothrin biodegradation by Bacillus thuringiensis strain ZS-19. Sci. Rep . 5:8784. doi: 10.1038/srep08784
Chen, S., Dong, Y. H., Chang, C., Deng, Y., Zhang, X. F., Zhong, G., et al. (2013). Characterization of a novel cyfluthrin-degrading bacterial strain Brevibacteriu maureum and its biochemical degradation pathway. Bioresour. Technol . 132, 16–23. doi: 10.1016/j.biortech.2013.01.002
Chen, S., Geng, P., Xiao, Y., and Hu, M. (2012). Bioremediation of β-cypermethrin and 3-phenoxybenzaldehyde contaminated soils using Streptomyces aureus HP-S-01. Appl. Microbiol. Biotechnol . 94, 505–515. doi: 10.1007/s00253-011-3640-5
Chen, S., and Zhan, H. (2019). “Biodegradation of synthetic pyrethroids insecticides,” in Microbial Metabolism of Xenobiotic Compounds , ed P. K. Arora (Springer Nature), 229–244. doi: 10.1007/978-981-13-7462-3_11
Chen, Z., Yin, H., Peng, H., Lu, G., Liu, Z., and Dang, Z. (2019). Identification of novel pathways for biotransformation of tertrabromobisphenol A by Phanerochaete chrysosporium combined with mechanism analysis at proteome level. Sci. Total Environ. 659, 1352–1362. doi: 10.1016/j.scitotenv.2018.12.446
Cheng, Y., Zang, H., Wang, H., Li, D., and Li, C. (2018). Global transcriptomic analysis of Rhocococcus erythopolis D310-1 in responding to chlorimuron-ethyl. Ecotoxicol. Environ. Saf. 157, 111–120. doi: 10.1016/j.ecoenv.2018.03.074
Colatriano, D., Ramachandran, A., Yergeau, E., Maranger, R., Gelinas, Y., and Walsh, D. A. (2015). Metaproteomics of aquatic microbial communities in a deep and stratified estuary. Proteomics 15, 3566–3579. doi: 10.1002/pmic.201500079
Colquhoun, D. R., Hartmann, E. M., and Halden, R. U. (2012). Proteomic profiling of the dioxin degrading bacterium Sphingomonas wittichii RW. J. Biomed. Biotechnol. 9:408690. doi: 10.1155/2012/408690
Cooper, L. A., Stringer, A. M., and Wade, J. T. (2018). Determining the specificity of cascade binding interference, and primed adaptation in-vivo in the Escherichia coli type IE CRISPR-Cas system. mBio 9:e02100-17. doi: 10.1128/mBio.02100-17
Crinnion, W. J. (2010). Toxic effects of the easily avoidable phthalates and parabens. Alternative Med. Rev. 15, 190–196.
PubMed Abstract | Google Scholar
Crump, B. C., Wojahn, J. M., Tomas, F., and Mueller, R. S. (2018). Metatranscriptomics and amplicon sequencing reveal mutualism in seagrass microiome. Front. Miocrobiol. 9:388. doi: 10.3389/fmicb.2018.00388
Cuozzo, S. A., Sineli, P. E., Davila Costa, J., and Tortella, G. (2017). Streptomyces sp. is a powerful biotechnological tool for the biodegradation of HCH isomers: biochemical and molecular basis. Crit. Rev. Biotechnol . 38, 719–728. doi: 10.1080/07388551.2017.1398133
Cycoń, M., Mrozik, A., and Piotrowska-Seget, Z. (2017). Bioaugmentation as a strategy for the remediation of pesticide-polluted soil: a review. Chemosphere 171, 52–71. doi: 10.1016/j.chemosphere.2016.12.129
Cycoń, M., and Piotrowska-Seget, Z. (2016). Pyrethroid-degrading microorganisms and their potential for the bioremediation of contaminated soils: a review. Front. Microbiol. 7:1463. doi: 10.3389/fmicb.2016.01463
Dangi, A. K., Sharma, B., Hill, R. T., and Shukla, P. (2018). Bioremediation through microbes: systems biology and metabolic engineering approach. Crit. Rev. Biotechnol. 39, 79–98. doi: 10.1080/07388551.2018.1500997
Delegan, Y. A., Valentovich, L. N., Shafieva, S. M., Ganbarov, K. G., Filonov, A. E., and Vainstein, M. B. (2019). Characterization and genome analysis of highly efficient thermotolerant oil-degrading bacterium Gordonia sp. 1D. Folia Microbiol. 64, 41–48. doi: 10.1007/s12223-018-0623-2
d'Errico, G., Aloj, V., Flematti, G. R., Sivasithamparam, K., and Worth, C. M. (2020). Metabolites of Drechslera sp. endophyte with potential as biocontrol and bioremediation agent. Nat. Prod. Res. 11, 1–9. doi: 10.1080/14786419.2020.1737058
Desai, C., Pathak, H., and Madawar, D. (2009). Advances in molecular and “omics” technologies to gauge microbial communities and bioremediation at xenobiotic/anthropogen contaminated sites. Bioresour. Technol. 101, 1558–1569. doi: 10.1016/j.biortech.2009.10.080
Devarapalli, P., and Kumavnath, R. N. (2015). “Metagenomics-a technological drift in bioremediation,” in Advances in Bioremediation of Wastewater and Polluted Soil , ed N. Shiomi (Rijeka: Intech Open), 73–91. doi: 10.5772/60749
Dhakal, K., Gadupadi, G. S., and Robertson, L. W. (2017). Sources and toxicities of phenolic polychlorinated biphenyls (OH-PCBs). PCBs Risk Eval. Environ. Protect. 25, 16277–16290. doi: 10.1007/s11356-017-9694-x
Diaz, E. (2004). Bacterial degradation of aromatic pollutants: a paradigm of metabolic versatility. Int. Microbiol. 7, 173–180.
Ding, J., Yu, Z., Wang, H., Jian, H., Leng, H., and Xiao, X. (2017). Microbial community structure of deep sea hydrothermal vents on the ultraslow spreading southwest Indian ridge. Front. Microbiol. 8:1012. doi: 10.3389/fmicb.2017.01012
Dinka, D. D. (2018). Environmental xenobiotics and their adverse health impacts-a general review. J. Environ. Poll. Human Health . 6, 77–88. doi: 10.12691/jephh-6-3-1
Dovrak, P., Nikel, P. I., Damborsky, J., and de Lorenzo, V. (2017). Bioremediation 3.0: engineering pollutant-removing bacteria in the times of systemic biology. Biotechnol. Adv. 35, 845–866. doi: 10.1016/j.biotechadv.2017.08.001
Durand, S., Sancelme, M., Besse-Hoggan, P., and Combourieu, B. (2010). Biodegradation pathway of mesotrione: complementaries of NMR, LC-NMR, and LC-MS for qualitative and quantitative metabolomic profiling. Chemosphere 81, 372–380. doi: 10.1016/j.chemosphere.2010.07.017
Dutta, T. K., Selifonov, S. A., and Gunsalus, I. C. (1998). Oxidation of methyl-substituted napthalenes: pathways in a versatile Sphingomonas paucimobilis strain. Appl. Environ. Microbiol. 64, 1884–1889. doi: 10.1128/AEM.64.5.1884-1889.1998
Ellis, L. B. M., Roe, D., and Wackett, L. P. (2006). The University of Minnesota biocatalyst/biodegradation database: the first decade. Nucleic Acid Res. 34, D517–D521. doi: 10.1093/nar/gkj076
Embrandiri, A., Kiyasudeen, S. K., Rupani, P. F., and Ibrahim, M. H. (2016). “Environmental xenobiotics and its effect on natural ecosystem”, in Plant Response to Xenobiotic , eds A. Singh, S. M. Prasad, and R. P. Singh (Singapore: Springer), 1–18. doi: 10.1007/978-981-10-2860-1_1
Endo, R., Kamakura, M., Miyauchi, K., Fukuda, M., Ohtsubo, Y., Tsuda, M., et al. (2005). Identification and characterization of genes involved in the downstream degradation pathway of γ-hexachlorocyclohexane in Sphingomonas paucimobilus UT26. J. Bacteriol . 187, 847–853. doi: 10.1128/JB.187.3.847-853.2005
Farnandez, M., Duque, E., Pizarro-Tobias, P., van Dillewijn, P., Wittich, R. M., and Ramos, J. L. (2009). Microbial responses to xenobiotic compounds. Identification of genes that allow Pseudomonas putida KT2440 to cope with 2,4,6-trinitrotoluene. Microb. Biotechnol . 2, 287–294. doi: 10.1111/j.1751-7915.2009.00085.x
Fida, T. T., Moreno-Forero, S. K., Breugelmans, P., Heipieper, H. J., Roling, W. F. M., and Springael, D. (2017). Physiological and transcriptome response of the polycyclic aromatic hydrocarbon degrading Novosphingobium sp. LH128 after inoculation in soil. Environ. Sci. Technol. 51, 1570–1579. doi: 10.1021/acs.est.6b03822
Franzosa, E. A., Hsu, T., Sirota-Madi, A., Shafquat, A., Abu-Ali, G., Morgan, X. C., et al. (2015). Sequencing and beyond: integrating molecular ‘omics for microbial community profiling. Nat. Rev. Microbiol. 13, 360–372. doi: 10.1038/nrmicro3451
French, K. E., Zhou, Z., and Terry, N. (2020). Horizontal ‘gene drives’ harness indigenous bacteria for bioremediation. Sci. Rep . 10:15091. doi: 10.1038/s41598-020-72138-9
Fuks, G., Elgert, M., Amir, A., Zeisel, A., Turnbaugh, P. J., Soen, Y., et al. (2018). Combining 16S rRNA gene variable regions enables high resolution microbial community profiling. Microbiome 6:17. doi: 10.1186/s40168-017-0396-x
Fulekar, M. H., and Geetha, M. (2008). Bioremediation of chloropyrifos by Pseudomonas aeruginosa using scale up technique. J. Appl. Biosci . 12, 657–660.
Gammon, D. W., Liu, Z., Chandrasekaran, A., El-Naggar, S. F., Kuryshev, Y. A., and Jackson, S. (2019). Pyrethroid neurotoxicity studies with bifenthrin indicate a mixed type I/II mode of action. Pest Manage. Sci. 75, 1190–1197. doi: 10.1002/ps.5300
Garrido-Sanz, D., Manzano, J., Martin, M., Redondo-Nieto, M., and Rivillia, R. (2018). Metagenomic analysis of a biphenyl-degrading soil bacterial consortium reveals the metabolic roles of specific populations. Front. Microbiol. 9:232. doi: 10.3389/fmicb.2018.00232
Geueke, B., Garg, N., Ghosh, S., Fleischmann, T., Holliger, C., Lal, R., et al. (2013). Metabolomics of hexachlorocyclohexane (HCH) transformation: ratio of LinA to LinB determines metabolic fate of HCH isomers. Environ. Microbiol. 15, 1040–1049. doi: 10.1111/1462-2920.12009
Ghosal, D., Ghosh, S., Dutta, T. K., and Ahn, Y. (2016). Current state of knowledge in microbial degradation of polycyclic aromatic hydrocarbons (PAHs): a review. Front. Microbiol. 7:1369. doi: 10.3389/fmicb.2016.01369
Gillbride, K. A., Lee, D. Y., and Beaudette, L. A. (2006). Molecular techniques in wastewater: understanding microbial communities, detecting pathogens, and real-time process control. J. Microbiol. Methods 66, 1–20. doi: 10.1016/j.mimet.2006.02.016
Gilliespie, I. M. M., and Philp, J. C. (2013). Bioremediation, an environmental remediation technology for the bioeconoomy. Trends Biotechnol. 31, 329–332. doi: 10.1016/j.tibtech.2013.01.015
Giri, K., Rawat, A. P., Rawat, M., and Rai, J. (2014). Biodegradation of hexachlorocyclohexane by two species of bacillus isolated from contaminated soil. Chem. Ecol . 30, 97–109. doi: 10.1080/02757540.2013.844795
Godheja, J., Shekhar, S. K., and Modi, D. R. (2014). Advances in molecular biology approaches to guage microbial communities and bioremediation at contaminated sites. Int. J. Environ. Bioremediate. Biodegrade. 2, 167–177. doi: 10.12691/ijebb-2-4-4
Godheja, J., Shekhar, S. K., Siddiqui, S. A., and Modi, D. R. (2016). Xenobiotic compounds present in soil and water: a review on remediation strategies. J. Environ. Anal. Toxicol . 6:2161. doi: 10.4172/2161-0525.1000392
Greene, A. C. (2018). CRISPR-based antibacterials: transforming bacterial defense into offense. Trends Biotechnol. 36, 127–130. doi: 10.1016/j.tibtech.2017.10.021
Griffiths, W. (2007). Metabolomics, Metabonomics and Metabolite Profiling. Cambridge: Royal Society of Chemistry. doi: 10.1039/9781847558107
Grob, C., Taubert, M., Howat, A. M., Burns, O. J., Dixon, J. L., Richnow, H. H., et al. (2015). Combining metagenomics with metaproteomics and stable isotope probing reveales metabolic pathways used by a naturally occurring marine methylotroph. Environ. Microbiol. 17, 4007–4018. doi: 10.1111/1462-2920.12935
Gu, Q., Wu, Q., Zhang, J., Guo, W., Ding, Y., Wang, J., et al. (2018). Isolation and transcriptome analysis of phenol-degrading bacterium from carbon-sand filters in full scale drinking water treatment plant. Front. Microbiol. 9:2162. doi: 10.3389/fmicb.2018.02162
Gursoy, S., and Can, M. (2019). Hypervariable regions in 16S rRNA genes for taxonomic classification. Se. Eur. J. Soft Comput. 8, 23–26. doi: 10.21533/scjournal.v8i1.171
Gutierrez, D. B., Gant-Branum, R. L., Romer, C. E., Farrow, M. A., Allen, J. L., Dahal, N., et al. (2018). An integrated, high-throughput strategy for “multi-omic” system level analysis. J. Proteome Res . 17, 3396–3408. doi: 10.1021/acs.jproteome.8b00302
Gutleben, J., Chaib De Mares, M., van Elsas, J. D., Smidt, H., Overmann, J., and Sipkema, D. (2018). The multi-omics promise in context: from sequence to microbial isolate. Crit. Rev. Microbiol . 44, 212–229. doi: 10.1080/1040841X.2017.1332003
Handelsman, J. (2004). Metagenomics: application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev . 68, 669–685. doi: 10.1128/MMBR.68.4.669-685.2004
Hashmi, M. Z., Kumar, V., and Verma, A. (2017). Xenobiotics in the Soil Environment: Monitoring, Toxicity and Management. Cham: Springer International Publishing. doi: 10.1007/978-3-319-47744-2
He, Y., Feng, X., Feng, J., Zhang, Y., and Xiao, X. (2015). Metagenome and metatrancriptome revealed a highly active and intensive sulfur cycle in an oil-immersed hydrothermal chimney in Guaymas Basin. Front. Microbiol. 6:1236. doi: 10.3389/fmicb.2015.01236
He, Z., Deng, Y., van Nostrand, J. D., Xu, M., Hemme, L. H., Tu, Q., et al. (2010). GeoChip 3.0 as a high-throughput tool for analyzing microbial community composition, structure and functional activity. ISME J. 4, 1167–1179. doi: 10.1038/ismej.2010.46
Hegedus, B., Kos, P. B., Bende, G., Bounedjoum, N., Maroti, G., Laczi, K., et al. (2018). Starvation and xenobiotic- related transcriptomic responses of the sulfanilic acid-degrading bacterium Novosphingobium resinvorum SA1. Appl. Microbiol. Biotechnol. 102, 305–318. doi: 10.1007/s00253-017-8553-5
Hettich, R. L., Pan, C., Chourey, K., and Giannone, R. J. (2013). Metaproteomics: Harnessing the power of high performance mass spectrometry to identify the suite of proteins that control metabolic activities in microbial communities. Anal. Chem . 85, 4203–4214. doi: 10.1021/ac303053e
Hidalgo, K. J., Teramoto, E. H., Soriano, A. U., Valoni, E., Baessa, M. P., Richnow, H. H., et al. (2020). Taxonomic and functional diversity of the microbiome in a jet fuel contaminated site as revealed by combined application of in situ microcosms with metagenomic analysis. Sci. Total Environ. 708:135152. doi: 10.1016/j.scitotenv.2019.135152
Hong, Y. H., Deng, M. C., Xu, X. M., Wu, C. F., Xiao, X., and Zhu, Q. (2016). Characterization of the transcriptome of Achromobacter sp. HZ01 with the outstanding hydrocarbon-degrading ability. Gene 584, 185–194. doi: 10.1016/j.gene.2016.02.032
Huang, Y., Lin, Z., Zhang, W., Pang, S., Bhatt, P., Rene, E. R., et al. (2020). New insights into the microbial degradation of D -cyphenothrin in contaminated water/soil environments. Microorganisms 8:473. doi: 10.3390/microorganisms8040473
Huang, Y., Zhang, W., Pang, S., Chen, J., Bhatt, P., Mishra, S., et al. (2021). Insights into the microbial degradation and catalytic mechanisms of chlorpyrifos. Environ. Res. 194:110660. doi: 10.1016/j.envres.2020.110660
Hussain, I., Aleti, G., Naidu, R., Puschenreiter, M., Mahmood, Q., and Rahman, M. M. (2018). Microbe and plant assisted remediation of organic xenobiotics and its enhancement by genetically modified organisms and recombinant technology: a review. Sci. Total Environ. 628, 1582–1599. doi: 10.1016/j.scitotenv.2018.02.037
Iovdijova, A., and Bencko, V. (2010). Potential risk of exposure to selected xenobiotic residues and their fate in the food chain-part I: classification of xenobiotics. Annl. Agric. Environ. Med. 17, 183–192.
Jacob, J., and Cherian, J. (2013). Review of environmental and human exposure to persistent organic pollutant. Asian Social Sci . 9:11. doi: 10.5539/ass.v9n11P107
Jagadeesh, D. S., Kannegundla, U., and Reddy, R. K. (2017). Application of proteomic tools in food quality and safety. Adv. Anim. Vet. Sci. 5, 213–225. doi: 10.17582/journal.aavs/2017/5.5.213.225
Jaiswal, S., and Shukla, P. (2020). Alternative strategies for microbial remediation of pollutants via synthetic biology. Front. Microbiol . 11:808. doi: 10.3389/fmicb.2020.00808
Jaiswal, S., Singh, D. K., and Shukla, P. (2019). Gene editing and systems biology tools for pesticide bioremediation: a review. Front. Microiol. 10:87. doi: 10.3389/fmicb.2019.00087
Jalowiecki, L., Chojniak, J. M., Dorgeloh, E., Hegedusova, B., Ejhed, H., Magner, J., et al. (2016). Microbial community profiles in wastewaters from onsite wastewater treatment system technology. PLoS ONE 11:e0147725. doi: 10.1371/journal.pone.0147725
Janssen, D. B., and Stucki, G. (2020). Perspectives of genetically engineered microbes for groundwater bioremediation. Environ. Sci. Proc. Imp. 22:487. doi: 10.1039/C9EM00601J
Jayaraj, R., Megha, P., and Sreedev, P. (2016). Organochlorine pesticides, their toxic effects on living organisms and their fate in the environment. Interdiscip. Toxicol. 9, 90–100. doi: 10.1515/intox-2016-0012
Jeffries, T. C., Rayu, S., Nielsen, U. N., Lai, K., Ijaz, A., Nazaries, L., et al. (2019). Metagenomics function potential predicts functional rates of a model organophosphorus xenobiotic in pesticide-contaminated soil. Front. Microbiol . 9:147. doi: 10.3389/fmicb.2018.00147
Jehmlich, N., Kleinsteuber, S., Vogt, C., Benndorf, D., Harms, H., Schmidt, F., et al. (2010). Phylogenetic and proteomic analysis of an anaerobic toluene-degrading community. J. Appl. Microbiol. 109, 1937–1945. doi: 10.1111/j.1365-2672.2010.04823.x
Jing, R., Fusi, S., and Kjellerup, B. V. (2018). Remediation of polychlorinated bisphenyls (PCBs) in contaminated soils and sediments: state of knowledge and perspectives. Front. Environ. Sci. 6:79. doi: 10.3389/fenvs.2018.00079
Johnson, C. H., Patterson, A. D., Idle, J. R., and Gonzalez, F. J. (2011). Xenobiotic metabolomics: major impact on the metabolome. Annu. Rev. Pharmacol. Toxicol. 52, 37–56. doi: 10.1146/annurev-pharmtox-010611-134748
Johnson, S. J., Spakowicz, D. J., Hong, B.-Y., Petersen, L., Demkowicz, P., Chen, L., et al. (2019). Evaluation of 16S rRNA gene sequencing for sepecies and strain-levl microbiome analysis. Nat. Commun. 10:5029. doi: 10.1038/s41467-019-13036-1
Jones, O. A. H., Sdepanian, S., Lofts, S., Svendsen, C., Spurgeon, D. J., Maguire, M. L., et al. (2014). Metabolomic analysis of soil communities can be used for pollution assessment. Environ. Toxicol. Chem. 33, 61–64. doi: 10.1002/etc.2418
Junghare, M., Spiteller, D., and Schink, B. (2019). Anaerobic degradation of xenobiotic isophthalate by the fermenting bacterium Syntrophorhabdus aromaticivorans. ISME J. 13, 1252–1258. doi: 10.1038/s41396-019-0348-5
Kale, N. S., Haug, K., Conesa, P., Jayseelam, K., Moreno, P., Rocca-Serra, P., et al. (2016). MetaboLights: an open-access database repository for metabolomics data. Curr. Protoc. Bioinform . 53:14. doi: 10.1002/0471250953.bi1413s53
Kanehisa, M., Furumichi, M., and Tanabe, M. (2017). KEGG: new perspectives on genome, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. doi: 10.1093/nar/gkw1092
Kaul, S., Sharma, T., and Dhar, M. K. (2016). “Omics” tools for better understanding the plant endophyte interactions. Front. Plant Sci. 7:955. doi: 10.3389/fpls.2016.00955
Kaur, H., and Kaur, G. (2016). Application of ligninolytic potentials of a white-rot fungus Ganoderma lucidum for degradation of lindane. Environ. Monit. Assess . 188:588. doi: 10.1007/s10661-016-5606-7
Keller, M., and Hettich, R. (2009). Environmental proteomics: a paradigm shift in characterizing microbial activities at the molecular level. Microbiol. Mol. Biol. Rev . 73, 62–70. doi: 10.1128/MMBR.00028-08
Kessner, D., Chambers, M., and Burke, R. (2008). ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536. doi: 10.1093/bioinformatics/btn323
Keum, Y. S., Seo, J. S., Li, Q. X., and Kim, J. H. (2008). Comparative metabolomic analysis of Sinorhizobium sp. C4 during the degradation of phenanthrene. Appl. Microbiol. Biotechnol . 80, 863–872. doi: 10.1007/s00253-008-1581-4
Kim, K. H., Jahan, S. A., Kabir, E., and Brown, R. C. B. (2013). A review of airborne polycyclic aromatic hydrocarbons (PAHs) and their human health effect. Environ. Int . 60, 71–80. doi: 10.1016/j.envint.2013.07.019
Kim, S. I., Song, S. Y., Kim, K. W., Ho, E. M., and Oh, K. H. (2003). Proteomic analysis of the benzoate degradation pathway in Acinetobacter sp. KS-1. Res. Microbiol. 10, 679–703. doi: 10.1016/j.resmic.2003.09.003
Kim, S. J., Jones, R. C., and Cha, C. J. (2004). Identification of proteins induced by polycyclic aromatic hydrocarbon in Mycobacterium vanbaalenii PYR-1 using two-dimensional polyacrylamide gel electrophoresis and denovo sequencing methods. Proteomics 4, 3899–3908. doi: 10.1002/pmic.200400872
Kiyohara, H., Torigoe, S., Kaida, N., Asaki, T., Iida, T., Hayashi, H., et al. (1994). Cloning and characterization of a chromosomal gene cluster, pah , that encodes the upper pathway for phenanthrene and naphthalene utilization by Pseudomonas putida OUS82. J. Bacteriol. 176, 2439–2443. doi: 10.1128/JB.176.8.2439-2443.1994
Knief, C. (2014). Analysis of plant microbe interactions in the era of next generation sequencing technologies. Front. Plant Sci. 5:216. doi: 10.3389/fpls.2014.00216
Krivobok, S., Kuony, S., Meyer, C., and Louwagie, M. (2003). Identification of pyrene-induced proteins in Mycobacterium sp. strain 6PY1: evidence for two-ring hydroxylating dioxygenases. J. Bacteriol. 185, 3828–3841. doi: 10.1128/JB.185.13.3828-3841.2003
Kumar, R., Kumar, D., Pandya, L., Pandit, P. R., Patel, Z., Bhairappanavar, S., et al. (2020). “Gene-targeted metagenomics approach for the degradation of organic pollutants,” in Emerging Technologies in Environmental Bioremediation , eds M. P. Shah, S. Rodriguez-Couto, and S. S. Sengor (Amsterdam: Elsevier), 257–273. doi: 10.1016/B978-0-12-819860-5.00010-9
Kumar, S. S., Shantkriti, S., Muruganandham, T., Murugesh, E., Rane, N., and Govindwar, S. P. (2016). Bioinformatics aided microbial approach for bioremediation of wastewater containing textile dyes. Ecol. Info. 31, 112–121. doi: 10.1016/j.ecoinf.2015.12.001
Kuzyakov, Y., and Blagodatskaya, E. (2015). Microbial hotspots and hot moments in soil, concept and review. Soil Biol. Biochem. 183, 184–199. doi: 10.1016/j.soilbio.2015.01.025
Lallas, P. L. (2001). The Stockholm convention on persistent organic pollutant. Am. J. Int. Law 95, 692–708. doi: 10.2307/2668517
Lam, K. N., Cheng, J., Engel, K., Neufeld, J. D., and Charles, T. C. (2015). Current and future resources for functional metagenomics. Front. Microbiol. 6:1196. doi: 10.3389/fmicb.2015.01196
Lee, S. Y., Kim, G. H., Yun, S. H., Choi, C. W., Yi, Y. S., and Kim, J. (2016a). Proteogenomic characterization of monocyclic aromatic hydrocarbon degrading bacterium Burkholderia sp. K24. PLoS ONE 11:e0154233. doi: 10.1371/journal.pone.0154233
Lee, S. Y., Sekhon, S. S., Ban, Y. H., Ahn, J. Y., Ko, J. H., Lee, L., et al. (2016b). Proteomic analysis of polycyclic aromatic hydrocarbons (PAHs) degradation and detoxification in Sphingobium chungbukense DJ77. J. Microbiol. Biotechnol. 26, 1943–1950. doi: 10.4014/jmb.1606.06005
Li, C., Ma, Y., Mi, Z., Huo, R., Zhou, T., Hai, H., et al. (2018). Screening for Lactobacillus plantarum strains that posses organophosphorus pesticide-degrading activity and metabolomic analysis of phorate degradation. Front. Microbiol . 9:2048. doi: 10.3389/fmicb.2018.02048
Li, J., Wu, C., Chen, S., Lu, Q., Shim, H., Huang, X., et al. (2020). Enriching indigenous microbial consortia as a promising strategy for xenobiotic's cleanup. J. Clean. Prod. 261:121234. doi: 10.1016/j.jclepro.2020.121234
Li, M., Jain, S., Baker, B. J., Taylor, C., and Dick, G. J. (2014). Novel hydrocarbon monooxygenase genes in the metatrancriptome of natural deep-sea hydrocarbon plume. Environ. Microbiol. 16, 60–71. doi: 10.1111/1462-2920.12182
Li, S. H., Yu, X. Y., and Park, D. J. (2015). Rhodococcus soli sp. nov., an actinobacterium isolated from soil using a resuscitative technique. Antonie Van Leeuwenhoek 107, 357–366. doi: 10.1007/s10482-014-0334-x
Li, X., Qu, C., Bian, Y., Gu, C., Jiang, X., and Song, Y. (2019). New insights into responses of soil microorganisms to polycyclic aromatic hydrocarbon stress by combining enzyme activity and sequencing analysis with metabolomics. Environ. Pollut. 255:113312. doi: 10.1016/j.envpol.2019.113312
Lima-Morales, D., Jauregui, R., Camarinha-Silva, A., Geffers, R., Pieper, D. H., and Vilchez-Vergas, R. (2016). Linking microbial community and catabolic gene structures during the adaptation of three contaminated soils under continuous long-term polluted stress. Appl. Environ. Microbiol. 82, 2227–2237. doi: 10.1128/AEM.03482-15
Lin, Z., Pang, S., Zhang, W., Mishra, S., Bhatt, P., and Chen, S. (2020). Degradation of acephate and its intermediate methamidophos: mechanisms and biochemical pathways. Front. Microbiol. 11:2045. doi: 10.3389/fmicb.2020.02045
Lindon, J. C., Nicholson, J. K., and Holmes, E. (2006). The Handbook of Metabonomics and Metabolomics . London: Elsevier Science.
Liu, L., Bilal, M., Duan, X., and Iqbal, H. M. N. (2019). Mitigation of environmental pollution by genetically engineered bacteria-current challenges and future perspectives. Sci. Total. Environ. 667, 444–454. doi: 10.1016/j.scitotenv.2019.02.390
Liu, L., Li, Y., and Li, S. (2012). Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012:251364. doi: 10.1155/2012/251364
Liu, S., Gu, C., Dang, Z., and Liang, X. (2017). Comparitive proteomics reveal the mechanism of Tween 80 enhanced phenantherene biodegradation by Sphingomonas sp. GY2B. Ecotoxicol. Environ. Saf. 137, 256–264. doi: 10.1016/j.ecoenv.2016.12.015
Lourenco, A., Ferreira, A., Veiga, N., Machado, I., Pereira, M. O., and Azevedo, N. F. (2012). BiofOmics: a webplatform for the systematic and standardized collection of high-throughput biofilmdata. PLoS ONE 7:e39960. doi: 10.1371/journal.pone.0039960
Lu, H., Giordano, F., and Ning, Z. (2016). Oxford nanopore MinION sequencing and genome assembly. Genom. Proteom. Bioinform. 14, 266–279. doi: 10.1016/j.gpb.2016.05.004
Lu, Z., Gan, J., Cui, X., Moreno, L. D., and Lin, K. (2019). Understanding the bioavailability of pyrethroids in the aquatic environment using chemical approaches. Environ. Int. 129, 194–207. doi: 10.1016/j.envint.2019.05.035
Lueders, T. (2015). “DNA-and RNA based stable isotope probing of hydrocarbon degraders”, in Hydrocarbon and Lipid Microbiology Protocols , eds T. J. McGenity, K. N. Timmis, and B. Nogales (New York, NY: Humana Press), 181–197. doi: 10.1007/8623_2015_74
Luo, C., Tsementzi, D., and Kyrpides, N. (2012). Direct comparison of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS ONE 7:e30087. doi: 10.1371/journal.pone.0030087
Machado, L., Leite, D. C. A., Rachid, C. T. C. C., Paes J. E., Martins, E., Peixoto, R., and Rosado, A. (2019). Tracking mangrove oil bioremediation approaches and bacterial diversity at different depths in an in situ mesocosms system. Front. Microbiol . 10:2107. doi: 10.3389/fmicb.2019.02107
Mahmoud, Y. A. G. (2016). Advancement in bioremediation process: a mini review. Int. J. Environ. Tech. Sci. 3, 83–94.
Malik, S., Beer, M., Megharaj, M., and Naidu, R. (2008). The use of molecular techniques to characterize the microbial communities in contaminated soil and water. Environ. Int. 34, 265–276. doi: 10.1016/j.envint.2007.09.001
Malla, M. A., Dubey, A., Yadav, S., Kumar, A., Hashem, A., and Abd-Allah, E. F. (2018). Understanding and designing the strategies for the microbe-mediated remediation of environmental contaminants using omics approaches. Front. Microbiol. 9:1132. doi: 10.3389/fmicb.2018.01132
Mallick, H., Franzosa, E. A., Mclver, L. J., Banerjee, S., Sirota-Madi, A., Kostic, A. D., et al. (2019). Predictive metabolomic profiling of microbial communities using amplicon or metagenomic sequences. Nat. Commun . 10:3136. doi: 10.1038/s41467-019-10927-1
Mao, X., Trembley, J., Yu, K., Tringe, S. G., and Alvarez-Cohen, L. (2019). Structural dynamics and transcriptomic analysis of Dehalococcoides mccartyi within a TCE-dechlorinating community in a completely mixed flow. Water Res. 158, 146–156. doi: 10.1016/j.watres.2019.04.038
Marcelino, V. R., Irinyi, L., Eden, J. S., Meyer, W., Holmes, E. C., and Sorrell, T. C. (2019). Metatranscriptomics as a tool to identify fungal species and subspecies in mixed communities- a proof of concept under laboratory conditions. IMA Fungus 10:12. doi: 10.1186/s43008-019-0012-8
Marco, D. E., and Abram, F. (2019). Editorial: using genomics, metagenomics and other “omics” to assess valuable microbial ecosystem services and novel biotechnological applications. Front. Microbiol. 10:151. doi: 10.3389/fmicb.2019.00151
Maurya, P. K. (2016). Bioaccumulation of xenobiotics compound of pesticides in riverine system and its control technique: a critical review. J. Ind. Poll. Control 32, 580–594.
Mazzoli, R., Pessione, E., Giuffrida, M. G., Fattori, P., Barello, C., Giunta, C., et al. (2007). Degradation of aromatic compounds by Acinetobacter radioresistens S13: growth characteristics on single substrates and mixtures. Arch. Microbiol. 188, 55–68. doi: 10.1007/s00203-007-0223-z
McGrath, K. C., Thomas-Hall, S. R., Cheng, C. T., Leo, L., Alexa, A., Schmidt, S., et al. (2008). Isolation and analysis of mRNA from environmental microbial communities. J. Microbiol. Methods 75, 172–176. doi: 10.1016/j.mimet.2008.05.019
Megharaj, M., and Naidu, R. (2010). Soil and brown field bioremediation. Microbial. Biotechnol. 10, 1244–1249. doi: 10.1111/1751-7915.12840
Mishra, S., Zhang, W., Lin, Z., Pang, S., Huang, Y., Bhatt, P., et al. (2020). Carbofuran toxicity and its microbial degradation in contaminated environments. Chemosphere 259:127429. doi: 10.1016/j.chemosphere.2020.127419
Mishra, V. K., Singh, G., and Shukla, R. (2019). Impact of xenobiotics under a changing climatic scenario. Clim. Change Agri. Ecosys. 2, 133–151. doi: 10.1016/B978-0-12-816483-9.00006-2
Misra, B. B., Langefeld, C. D., Olivier, M., and Cox, L.A. (2018). Integrated omics: tools, advances and future approaches. J. Mol. Endocrinol. 62, R21–R45. doi: 10.1530/JME-18-0055
Mocalli, S., and Benedetti, A. (2010). Exploring research frontiers in microbiology: the challenge of metagenomics in soil microbiology. Res. Microbiol. 161, 497–505. doi: 10.1016/j.resmic.2010.04.010
Moniruzzaman, M., Wurch, L. L., Alexander, H., Dyhrman, S. T., Gobler, C. J., and Wilhelm, S. W. (2017). Virus-host relationships of marine single-celled eukaryotes resolved from metatranscriptomics. Nat. Commun. 8:16054. doi: 10.1038/ncomms16054
Moody, J. D., Freeman, J. P., and Cerniglia, C. E. (2005). Degradation of benz[a]anthracene by Microbacterium vanbaalenii strain PYR-1. Biodegradation 16, 513–526. doi: 10.1007/s10532-004-7217-1
Moriya, Y., Shigemizu, D., Hattori, M., Tokimatsu, T., Kotera, M., Goto, S., et al. (2010). Pathpred: an enzyme-catalyzed metabolic pathway prediction server. Nucleic Acid Res . 38, W138–W143. doi: 10.1093/nar/gkq318
Nagata, Y., Kato, H., Ostsubo, Y., and Tsuda, M. (2019). Lessons from the genomes of lindane-degradation sphingomonads. Environ. Microbiol. Rep . 11, 630–644. doi: 10.1111/1758-2229.12762
Nahurira, R., Ren, I., and Song, J. (2017). Degradation of di(2-ethylhexyl) phthalate by a novel Gordonia alkanivorans strain yc-r12. Curr. Microbiol. 74, 1–11. doi: 10.1007/s00284-016-1159-9
Nakano, K., Shiroma, A., Shimoji, M., Tamotsu, H., Ashimine, N., Ohki, S., et al. (2017). Advantages of genome sequencing by long read sequencer using SMRT technology in medical area. Hum. Cell 30, 149–161. doi: 10.1007/s13577-017-0168-8
Nascimento, F. X., Hernandez, G., Glick, B. R., and Rossi, M. J. (2020). Plant growth-promoting activities and genomic analysis of the stress resistant Bacillus megaterium STB1, a bacterium of agriculture and biotechnological interest. Biotenol. Rep. 25:e00406. doi: 10.1016/j.btre.2019.e00406
Negi, G., Gangola, S., Khati, P., Kumar, G., Srivastava, A., and Sharma, A. (2016). Differential expression and characterization of cypermethrin-degrading potential proteins in Bacillus thuringiensis strain SG4. 3 Biotech 6:225. doi: 10.1007/s13205-016-0541-4
Negi, V., and Lal, R. (2017). Metagenomic analysis of a complex community present in pond sediment. J. Genomics 5, 36–47. doi: 10.7150/jgen.16685
Ngara, T. R., and Zhang, H. (2018). Recent advances in function-based metagenomic screening. Genom. Proteom. Bioinform. 6, 405–415. doi: 10.1016/j.gpb.2018.01.002
Niu, J., Rang, Z., and Zhang, C. (2016). The succession pattern of soil microbial communities and its relationship with tobacco bacterial wilt. BMC Microbiol. 16:233. doi: 10.1186/s12866-016-0845-x
Nkongolo, K. K., and Kotha, R. N. (2020). Advances in monitoring soil microbial community dynamic and function. J. Appl. Genet. 61, 249–263. doi: 10.1007/s13353-020-00549-5
Nzila, A. (2013). Update on the catabolism of organic pollutant by bacteria. Environ. Pollut. 178, 474–482. doi: 10.1016/j.envpol.2013.03.042
Nzila, A., Ramirez, C. O., Musa, M. M., Sankara, S., Basheer, C., and Li, Q. X. (2018). Pyrene biodegradation and proteomic analysis in Achromobacter xylosoxidans , PY4 strain. Int. Biodeterior. Biodegrad. 175, 1294–1305. doi: 10.1016/j.ibiod.2018.03.014
Oliver, J. D. (2010). Recent findings on the viable but nonculturable stage in pathogenic bacteria. FEMS Microbiol. Rev. 34, 415–425. doi: 10.1111/j.1574-6976.2009.00200.x
Oliver, J. D., Dagher, M., and Linden, K. (2005). Induction of Escherichia coli and Salmonella typhimuriam into the viable but nonculturable state following chlorination of wastewater. J.Water Health 3, 249–257. doi: 10.2166/wh.2005.040
Ortega-Gonzalez, D. K., Martinez-Gonzalez, G., Flores, C. M., Zaragoza, D., Cancino-Diaz, J. C., Cruz-Maya, J. A., et al. (2015). Amycolatopsis sp. Poz14 isolated from oil-contaminated soil degrades polycyclic aromatic hydrocarbons. Int. Biodeterior. Biodegrad. 99, 165–173. doi: 10.1016/j.ibiod.2015.01.008
Ortiz, H. M. L., Salinas, E. S., Gonzalez, E. D., and Godnez, M. L. C. (2013). “Pesticide biodegradation: mechanisms, genetics and strategies to enhance the process”, in Biodegradation-Life of Science , eds R. Chamy and F. Rosenkranz (Rijeka: InTech Open), 251–287.
Ortiz-Hernandez, M. L., Castrejon-Godinez, M. L., Popoca-Ursino, E. C., and Cervantes-Decasac, F. R. (2018). “Strategies for biodegradation and bioremediation of pesticides in the environment,” in Strategies for Bioremediation of Organic and Inorganic Pollutants , eds M. S. Fuentes, V. L. Colin, and J. M. Saez (Boca Raton, FL: CRC Press), 95–115.
Pandey, A., Tripathi, P. H., Tripathi, A. H., Pandey, S. C., and Gangola, S. (2019). “Omics technology to study bioremediation and respective enzymes,” in Smart Bioremediation Technologies: Microbial Enzymes , ed P. Bhatt (Academic Press), 23–43. doi: 10.1016/B978-0-12-818307-6.00002-0
Pandey, R., Masood, F., Singh, H. P., and Batish, D. R. (2017). Polycyclic aromatic hydrocarbons as environmental pollutants: a review. Int. J. Adv. Res. Sci. Eng. 6, 1361–1369.
Pang, S., Lin, Z., Zhang, W., Mishra, S., Bhatt, P., and Chen, S. (2020). Insights into the microbial degradation and biochemical mechanisms of neonicotinoids. Front. Microbiol. 11, 868. doi: 10.3389/fmicb.2020.00868
Panigrahi, S., Velraj, P., and Rao, T. S. (2019). “Functional microbial diversity in contaminated environment and application in bioremediation,” in Microbial Diversity in the Genomic Era , eds S. Das and H. Dask (Elsevier; Academia Press), 349–385. doi: 10.1016/B978-0-12-814849-5.00021-6
Parisi, V. A., Brubaker, G. R., Zenker, M. J., Prince, R. C., Gieg, L. M., Da Silva, M. L., et al. (2009). Field metabolomics and laboratory assessments of anaerobic intrinsic bioremediation of hydrocarbons at a petroleum-contaminated site. Microbial. Biotechnol. 2, 202–212. doi: 10.1111/j.1751-7915.2009.00077.x
Pathiraja, G., Egodawatta, P., Goonetilleke, A., and Junior Te'o, V. S. (2019). Solubilization and degradation of polychlorinated biphenyls (PCBs) by naturally occurring facultative bacteria. Sci. Total Environ. 651, 2197–2207. doi: 10.1016/j.scitotenv.2018.10.127
Patowary, K., Patowary, R., Kalita, M. C., and Deka, S. (2016). Development of an efficient bacterial consortium for the potential remediation of hydrocarbons from contaminated sites. Front. Microbiol. 7:1092. doi: 10.3389/fmicb.2016.01092
Paul, D., Pandey, G., Pandey, J., and Jain, R. K. (2005). Accessing microbial diversity for bioremediation and environmental pollutant restoration. Trends Biotechnol. 23, 135–142. doi: 10.1016/j.tibtech.2005.01.001
Pazos, F., Guijas, D., Valencia, A., and De Lorenzo, V. (2005). MetaRouter: bioinformatics for bioremediation. Nucleic Acids Res. 45, D588–D592. doi: 10.1093/nar/gki068
Pedersen, J. A., Yeager, M. A., and Suffer, I. H. (2003). Xenobiotic organic compounds in runoff from fields irrigated with treated wastewater. J. Agric. Food Chem. 51, 1360–1372. doi: 10.1021/jf025953q
Perelo, L. W. (2010). In situ and bioremediation of organic pollutants in aquatic sediments. J. Hazard. Mater. 177, 81–89. doi: 10.1016/j.jhazmat.2009.12.090
Perez-Losada, M., Castro-Nallar, E., Bendall, M. L., Freishtat, R. J., and Crandall, K. A. (2015). Dual transcriptomic profiling of host and microbiota during health and disease in pediatric asthma. PLoS ONE 10:e0131819. doi: 10.1371/journal.pone.0131819
Perruchon, C., Vasileiadis, S., Rousidou, C., Papadopoulou, E. S., Tanou, G., Samiotaki, M., et al. (2017). Metabolic and cell adaptation mechanism revealed through genomic, proteomic and transcription analysis of a Sphingomonas haloaromaticamans strain degrading ortho-phenylphenol. Sci. Rep. 7:6449. doi: 10.1038/s41598-017-06727-6
Phale, P. S., Sharma, A., and Gautam, K. (2019). “Microbial degradation of xenobiotics like aromatic pollutants from the terrestrial environments”, in Pharmaceutical and Personal Care Products: Waste Management and Treatment Technology Emerging Contaminants and Micro Pollutants , eds M. N. V. Prasad, M. Vithanage, and A. Kapley (Elsevier), 259–278. doi: 10.1016/B978-0-12-816189-0.00011-1
Pinu, F. R., Beale, D. J., Paten, A. M., Kouremenos, K., Swarup, S., Schirra, H. J., et al. (2019). Systems biology and multi-omics integration: viewpoints from the metabolomics research community. Metabolites 9:76. doi: 10.3390/metabo9040076
Przybylinska, P. A., and Wyszkowski, M. (2016). Environmental contamination with phthalates and its impact on living organisms. Ecol. Chem. Eng. S. 23, 347–356. doi: 10.1515/eces-2016-0024
Puckett, S. P., Samples, R. M., Schloss, P. D., and Balunas, M. J. (2020). “Metabolomics and the microbiome: characterizing molecular diversity in complex microbial communities,” in Comprehensive Natural Products III: Chemistry and Biology , eds H. W. Liu and T. P. Begley (Elsevier), 502–518. doi: 10.1016/B978-0-12-409547-2.14802-4
Rabausch, U., Juergensen, J., Ilmberger, N., Bohnke, S., Fischer, S., Schubach, B., et al. (2013). Functional screening of metagenome and genome libraries for detection of novel flavonoid modifying enzymes. Appl. Environ. Microbiol. 79, 4551–4563. doi: 10.1128/AEM.01077-13
Rahimi, T., Niazi, A., Deihimi, T., Taghavi, S. M., Avatollahi, S., and Ebrahimie, E. (2018). Genome annotation and comparative genomic analysis of Bacillus subtilis MJ01 a new biodegradation strain isolated from oil contaminated soil. Funct. Integr. Genomics 18, 533–543. doi: 10.1007/s10142-018-0604-1
Ram, R. J., Verberkmoes, N. C., and Thelen, M. P. (2005). Community proteomics of a natural microbial biofilms. Science 308, 1915–1920. doi: 10.1126/science.1109070
Rastogi, G., and Sani, R. K. (2011). “Molecular techniques to assess microbial community structure, function and dynamics in the environment,” in Microbes and Microbial Technology , eds I. Ahmed, F. Ahmed, and J. Pichtel (New York, NY: Springer Nature), 29–57. doi: 10.1007/978-1-4419-7931-5_2
Ravindra, A. P., and Haq, S. A. (2019). “Effects of xenobiotics and their biodegradation in marine life,” in Smart Bioremediation Technologies: Microbial Enzymes , ed P. Bhatt (Academic Press), 63–81. doi: 10.1016/B978-0-12-818307-6.00004-4
Reena, R., Majhi, M. C., Arya, A. K., Kumar, R., and Kumar, A. (2012). BioRadBase: a database for bioremediation of radioactive waste. Afr. J. Biotechnol. 11, 8718–8721. doi: 10.5897/AJB12.020
Rodriguez, A., Castrejon-Godinez, M. L., Salazar-Bustamante, E., Gama-Martinez, Y., Sanchez-Salinas, E., Mussali-Galante, P., et al. (2020). Omics approaches to pesticide biodegradation. Curr. Microbiol. 77, 545–563. doi: 10.1007/s00284-020-01916-5
Roh, S. W., Abell, G., Kim, K. H., Nam, Y. D., and Bae, J. W. (2010). Comparing microarray and next generation sequencing technologies for microbial ecology research. Trends Biotechnol . 28, 291–299. doi: 10.1016/j.tibtech.2010.03.001
Rossello-Mora, R., and Amann, R. (2001). The species concept for prokaryotes. FEMS Microbiol. Rev . 25, 39–67. doi: 10.1016/S0168-6445(00)00040-1
Rowdhwal, S. S. S., and Chen, J. (2018). Toxic effect of di-2-ethylhexyl phthalate: an overview. BioMed Res. Int. 2:1750368. doi: 10.1155/2018/1750368
Sajid, M., Basheer, C., Narasimhan, K., Buhmeida, A., Qahtani, M. A., and Al-Ahwal, M. S. (2015). Persistent and endocrine disrupting organic pollutant: advancements and challenges in analysis, health concerns and clinical correlates. Nat. Environ. Poll. Tech. 15, 733–746.
Salam, M., and Verma, A. (2019). Bacterial community structure in soils contaminated with electronic waste pollutants from Delhi NCR, India. Electronic J. Biotechnol. 41, 72–80. doi: 10.1016/j.ejbt.2019.07.003
Salipante, S. J., Kawasahima, T., and Rosenthal, C. (2014). Performance comparison of Illumina and ion torrent next generation sequencing platforms for 16S rRNA-based bacterial community profiling. Appl. Environ. Microbiol. 80, 7583–7591. doi: 10.1128/AEM.02206-14
Sathishkumar, M., Binupriya, A. R., Balk, S., and Yun, S. (2008). Biodegradation of crude oil by individual bacterial strains and mixed bacterial consortium isolated from hydrocarbon contaminated areas. Clean 36, 92–96. doi: 10.1002/clen.200700042
Sato, Y., Hori, T., Koike, H., Navarro, R. R., Ogata, A., and Habe, H. (2019). Transcriptome analysis of activated sludge microbiomes reveals an unexpected role of minority nitrifiers in carbon metabolism. Comm. Biol. 2:179. doi: 10.1038/s42003-019-0418-2
Sayler, G. S., and Ripp, S. (2000). Field application of genetically engineered microorganisms for bioremediation processes. Curr. Opin. Biotechnol. 480, 1–9. doi: 10.1016/S0958-1669(00)00097-5
Schecter, A., Birnbaum, H., Ryan, J. J., and Constable, J. D. (2006). Dioxins: an overview. Environ. Res. 101, 419–428. doi: 10.1016/j.envres.2005.12.003
Scholer, A., Jacquiod, S., and Vestergaard, G. (2017). Analysis of soil microbial communities based on amplicons sequencing of marker genes. Biol. Fertil. Soils 53, 485–489. doi: 10.1007/s00374-017-1205-1
Segata, N., Boernigen, D., Tickle, T. L., Morgan, X. C., Garrett, W. S., and Huttenhower, C. (2013). Computational meta'omics for microbial community studies. Mol. Sys. Biol . 9:666. doi: 10.1038/msb.2013.22
Sengupta, K., Swain, M. T., Livingstone, P. G., Whiteworth, D. E., and Saha, P. (2019). Genome sequencing and comparative transcriptomics provide holistic view of 4-nitrophenol degradation and concurrent fatty acid catabolism by Rhodococcus sp. strain BUPNP1. Front. Microbiol. 9:3209. doi: 10.3389/fmicb.2018.03209
Seo, J., Keum, Y. S., and Li, Q. X. (2013). Metabolomic and proteomic insights into carbaryl catabolism by Burkholderia sp. C3 and degradation of ten N -methylcarbamates. Biodegradation 24, 795–811. doi: 10.1007/s10532-013-9629-2
Seo, J. S., Keum, Y. S., and Li, Q. X. (2009). Bacterial degradation of aromatic compounds. Int. J. Environ. Res. Public Health 6, 278–309. doi: 10.3390/ijerph6010278
Shah, V., Jain, K., Desai, C., and Madamwar, D. (2011). “Metagenomics and integrative-omics technologies in microbial bioremediation: current trends and potential applications”, in Metagenomics: Current Innovations and Future Trends , ed D. Marco (Norfolk: Caister Academic Press), 211–240.
Shakya, M., Lo, C. C., and Chain, P. S. G. (2019). Advances and challenges in metatranscriptomic analysis. Front. Genet. 10:904. doi: 10.3389/fgene.2019.00904
Shanker, J., Abhilash, P. C., Singh, H. B., Singh, R. P., and Singh, D. P. (2011). Genetically engineered bacteria: an emerging tool for environmental remediation and future perspectives. Gene 480, 1–9. doi: 10.1016/j.gene.2011.03.001
Shapiro, R. S., Chavez, A., and Collins, J. J. (2018). CRISPR-based genomic tools for the manipulation ofgenetically intractable microorganism. Nat. Rev. Microbiol. 16, 333–339. doi: 10.1038/s41579-018-0002-7
Sharma, A., Yadav, B., Rohatgi, S., and Yadav, B. (2018). Cypermethrin toxicity: a review. J. Forensic Sci. Criminal Inves. 9:555767. doi: 10.19080/JFSCI.2018.09.555767
Sharma, P. K., Sharma, V., Sharma, A., Bhatia, G., Singh, K., and Sharma, R. (2019). Comparative metatranscriptome analysis revealed broad response of microbial communities in two soil types, agriculture versus organic soil. J. Genet. Eng. Biotechnol. 17:6. doi: 10.1186/s43141-019-0006-3
Shekhar, S. K., Godheja, J., and Modi, D. R. (2020). “Molecular technologies for assessment of bioremediation and characterization of microbial communities of pollutant-contaminated sites” in, Bioremediation of Industrial Waste for Environmental Safety , eds. R. N. Bharagava and G. Saxena (Springer Nature, Singapore), 447–474. doi: 10.1007/978-981-13-3426-9_18
Shi, S., Nuccio, E., Herman, D. J., Rijkers, R., Estera, K., Li, J., et al. (2015). Succesional trajectories of rhizosphere bacterial communities over consecutive seasons. mBio 6, e00746-15. doi: 10.1128/mBio.00746-15
Shokralla, S., Gibson, J. F., and Niknakht, H. (2014). Nest-generation DNA barcoding: using next generation sequencing to enhance and accelerate DNA barcode capture from single specimen. Mol. Ecol. Resour. 14, 892–901. doi: 10.1111/1755-0998.12236
Siles, J. A., and Margesin, R. (2018). Insights into microbial communities mediating the bioremediation of hydrocarbon-contaminated soil from Alpine former military site. Appl. Microbiol. Biotechnol. 102, 4409–4421. doi: 10.1007/s00253-018-8932-6
Singh, A., Chaudhary, S., Dubey, B., and Prasad, V. (2016). “Microbial-mediated management of organic xenobiotic pollutants in agricultural lands”, in Plant Response to Xenobiotics , eds A. Singh, S. M. Prasad, and R. P. Singh (Singapore: Springer), 211–230. doi: 10.1007/978-981-10-2860-1_9
Singh, A. K., Tiwari, M. N., Prakash, O., and Singh, M. P. (2012). A current review of cypermethrin-induced neurotoxicity and nigrostriatal dopaminergic neurodegeneration. Curr. Neuropharmacol . 10, 64–71. doi: 10.2174/157015912799362779
Singh, D. P., Prabha, R., Gupta, V. K., and Verma, M. K. (2018). Metatranscriptomic analysis deciphers multifunctionl genes and enzymes linked with the degradation of aromatic compounds and pesticide in the wheat rhizosphere. Front. Microbiol. 9:1331. doi: 10.3389/fmicb.2018.01331
Singh, O. V. (2006). Proteomics and metabolomics: the molecular make-up of toxic aromatic pollutant bioremediation. Proteomics 6, 5481–5492. doi: 10.1002/pmic.200600200
Singh, O. V., and Nagaraj, N. S. (2006). Transcriptomics, proteomics and interactions: unique approaches to track the insights of bioremediation. Brief. Funct. Genomics Proteomics 4, 355–362. doi: 10.1093/bfgp/eli006
Singh, S., and Li, S. S. L. (2011). Phthalates: toxicological and inferred human diseases. Genomics 97, 148–157. doi: 10.1016/j.ygeno.2010.11.008
Singh, V., Gohil, N., Ramírez García, R., Braddick, D., and Fofié, C. K. (2018). Recent advances in CRISPR-Cas9 genome editing technology for biological and biomedical investigations. J. Cell. Biochem. 119, 81–94. doi: 10.1002/jcb.26165
Song, Y., Li, X., Yao, S., Yang, X., and Jiang, X. (2020). Correlations between soil metabolomics and bacterial community structures in the pepper rhizosphere under plastic greenhouse cultivation. Sci. Total Environ . 728:138439. doi: 10.1016/j.scitotenv.2020.138439
Steele, H. L., Jaeger, K. E., Daniel, R., and Streit, W. R. (2009). Advances in recovery of novel biocatalysts from metagenomes. J. Mol. Microbiol. Biotechnol. 16, 25–37. doi: 10.1159/000142892
Stein, H. P., Navajas-Perez, R., and Aranda, E. (2018). “Potential for CRISPR genetic engineering to increase xenobiotic degradation capacities in model fungi”, in Approaches in Bioremediation, Nanotechonology , eds R. Prasad and E. Aranda (Cham: Springer Nature), 61–78. doi: 10.1007/978-3-030-02369-0_4
Su, X., Chen, X., and Hu, J. (2013). Exploring the potential environmental functions of viable but non-culturable bacteria. World J. Biotechnol. 29, 2213–2218. doi: 10.1007/s11274-013-1390-5
Sun, G., Du, Y., Yin, J., Jiang, Y., Zhang, D., Jiang, B., et al. (2019). Response of microbial communities to different organochlorine pesticide (OCPs) contamination levels in contaminated soil. Chemosphere 215, 461–469. doi: 10.1016/j.chemosphere.2018.09.160
Sunita, V. J., Dolly, P. R., Bateja, S., and Vivek, U. N. (2013). Isolation and screening for hydrocarbon utilizing bacteria (HUB) from petroleum samples. Int. J. Curr. Appl. Sci. 2, 48–60.
Surani, J. J., Akbari, V. G., Purohit, M. K., and Singh, S. P. (2011). Pahbase, a freely available functional database of polycyclic aromatic hydrocarbons (Pahs) degrading bacteria. J. Bioremed. Biodegrad. 2, 116–135. doi: 10.4172/2155-6199.1000116
Szewczyk, R., Kusmierska, A., and Bernat, P. (2017). Ametryn removal by Metarhizium brunneum : biodegradation pathway proposal and metabolic background revealed. Chemosphere 190, 174–183. doi: 10.1016/j.chemosphere.2017.10.011
Szewczyk, R., Sobon, A., Sylwia, R., Dzitko, K., Waidelich, D., and Dlugonski, J. (2014). Intracellukar proteome expression during 4-n-nonylphenol biodegradation by filamentous fungus Metarhizium robertsii . Int. Biodeterior. Biodegrad. 93, 44–53. doi: 10.1016/j.ibiod.2014.04.026
Taupp, M., Mewis, K., and Hallam, S. J. (2011). The art and design of functional metagenomic screens. Curr. Opin. Biotechnol. 22, 465–472. doi: 10.1016/j.copbio.2011.02.010
Temperton, B., and Giovannoni, S. J. (2012). Metagenomics: microbial diversity through a scratched lens. Curr. Opin. Microbiol. 15, 605–612. doi: 10.1016/j.mib.2012.07.001
Theriot, C. M., Koenigsknecht, M. J., Carlson, P. E., Hatton, G. E., Nelson, A. M., Li, B., et al. (2014). Antibiotic-induced shifts in the mouse gut microbiome and metabolome increases susceptibility to Clostridium difficile infection. Nat. Commun. 5:3114. doi: 10.1038/ncomms4114
Tsaboula, A., Papadakis, E. N., Vrvzas, Z., and Kotopoulou, A. (2016). Environmental and human risk hierarchy of pesticide: a monitoring, hazard assessment and environmental fate. Environ. Int. 78–93. doi: 10.1016/j.envint.2016.02.008
Ullah, S., Zuberi, A., Alagawany, M., Farag, M. R., Dadar, M., Karthik, K., et al. (2018). Cypermethrin induced toxicities in fish and adverse health outcomes: its prevention and control measure adaptation. J. Environ. Manage. 206, 863–871. doi: 10.1016/j.jenvman.2017.11.076
Urbance, J. W., Cole, J., and Saxman, P. (2003). BSD: the biodegradative strain database. Nucleic Acids Res. 31, 152–155. doi: 10.1093/nar/gkg032
Vandera, E., Samiotaki, A., Parapouli, M., Panayotou, G., and Koukkou, A. I. (2015). Comparative proteomic analysis of Arthrobacter phenanthrenivorans Sphe3 on phenanthrene, Phthalate and glucose. J. Proteomic 115, 73–89. doi: 10.1016/j.jprot.2014.08.018
Velmurgan, N., Lee, H., Cha, H. J., and Lee, Y. S. (2017). Proteomic analysis of the marine-derived fungus Paecilomyces sp. strain SF-8 in response to polycyclic aromatic hydrocarbons. Botanica Mar. 60:101. doi: 10.1515/bot-2016-0101
Vizcaino, J. A., Csordas, A., del-Toro, N., Dianes, J. A., Griss, J., Lavidas, I., et al. (2016). Update of the PRIDE database and its related tools. Nucleic Acids Res . 44, D447–D456. doi: 10.1093/nar/gkw880
Wagner, J., Coupland, P., Browne, H. P., Lawley, T. D., Francis, S. C., and Parkhill, J. (2016). Evaluation of PacBio sequencing for full length bacterial 16S rRNA gene classification. BMC Microbiol. 16:274. doi: 10.1186/s12866-016-0891-4
Wang, D. Z., Kong, L. F., Li, Y. Y., and Xie, Z. X. (2016). Environmental microbial community proteomics: status, challenges and perspectives. Int. J. Mol. Sci. 17:1275. doi: 10.3390/ijms17081275
Wang, T., Hu, C., Zhang, R., Sun, A., Li, D., and Shi, X. (2019). Mechanism study of cyfluthrin biodegradation by Photobacterium ganghwense with comparative metabolomics. Appl. Microbiol. Biotechnol. 103, 473–488. doi: 10.1007/s00253-018-9458-7
Wang, W., Wang, L., and Shao, Z. (2018). Polycyclic aromatic hydrocarbon (PAH) degradation pathways of the obligate marine PAH degrader Cycloclasticus sp. strain P1. Appl. Environ. Microbiol. 84:e01261-18. doi: 10.1128/AEM.01261-18
Wang, Y. F., Zhu, H. W., Wang, Y., Zhang, X. L., and Tam, N. F. Y. (2018). Diversity and dynamics of microbial community structure in different mangrove, marine and freshwater sediments during anaerobic debromination of PBDEs. Front. Microiol. 9:952. doi: 10.3389/fmicb.2018.00952
Wei, K., Yin, H., Peng, H., Liu, Z., Lu, G., and Dang, Z. (2017). Characteristics and proteomic analysis of pyrene degradation by Brevibacillus brevis in liquid medium. Chemosphere 178, 80–87. doi: 10.1016/j.chemosphere.2017.03.049
White, R. A., Bottos, E. M., Roy, C. T., Zucker, J. D., Brislawn, C. J., Nicora, C. D., et al. (2016). Moleculo long-read sequencing facilitates assembly and genomic binning from complex soil metagenomes. mSystems 1:e00045-16. doi: 10.1128/mSystems.00045-16
Widada, J., Nojiri, H., and Omori, H. (2002). Recent developments in molecular techniques for identification and monitoring of xenobiotic-degrdaing bacteria and their catabolic genes in bioremediation. Appl. Microbiol. Biotechnol. 60, 45–59. doi: 10.1007/s00253-002-1072-y
Williams, T. J., Wilkins, D., Long, E., Evans, F., DeMaere, M. Z., Raftery, M. J., et al. (2013). The role of planktonic Flavobacteria in processing algal organic matter in coastal East Antartica revealed using metagenomics and metaproteomics. Environ. Microbiol . 15, 1302–1317. doi: 10.1111/1462-2920.12017
Withers, E., Hill, P. W., Chadwick, D. R., and Jones, D. L. (2020). Use of untargeted metabolomics for assessing soil quality and microbial function. Soil Biol. Biochem. 143:107758. doi: 10.1016/j.soilbio.2020.107758
Wolf, D. C., Cryder, Z., Khoury, R., Carlan, C., and Gan, J. (2020). Bioremediation of PAH-contaminated shooting range soil using integrated approaches. Sci. Total Environ. 726:13844. doi: 10.1016/j.scitotenv.2020.138440
Wong, D. W. S. (2018). “Gene targeting and genome editing,” in The ABCs of Gene Cloning , ed D. W. S. Wong (Cham: Springer), 187–197. doi: 10.1007/978-3-319-77982-9_20
Wright, R., Bosch, R., Gibson, M. I., and Christie-Oleza, J. (2020). Plasticizer degradation by a marine bacterial isolates: a proteogenomic and metabolomic characterization. Environ. Sci. Technol. 54, 2244–2256. doi: 10.1021/acs.est.9b05228
Wu, X., Yin, Y., Wang, S., and Yu, Y. (2014). Accumulation of chlorothalonil and its metabolite, 4-hydroxychlorothalonil in soil after repeated applications and its effects on soil microbial activities underground greenhouse conditions. Environ. Sci. Pollut. Res. 21, 3452–3459. doi: 10.1007/s11356-013-2318-1
Xie, J., He, Z., Liu, X., Van Nostrand, J. D., and Deng, Y. (2011). GeoChip based analysis of functional gene diversity and metabolic potential of microbial communities in acid mine drainage. Appl. Environ. Microbiol. 77, 991–999. doi: 10.1128/AEM.01798-10
Xiong, J. B., Wu, L. Y., Tu, S. X., Van Nostrand, J. D., He, Z. H., Zhou, J. Z., et al. (2010). Microbial communities and functional genes associated with soil arsenic contamination and the rhizosphere of arsenic-hyperaccumulating plant Pteris vittata L. Appl. Environ. Microbiol. 76, 7277–7284. doi: 10.1128/AEM.00500-10
Xu, Z., Hansen, M. A., Hansen, L. H., Jacquiod, S., and Soresen, S. J. (2014). Bioinformatic approaches reveal metagenomic characterization of soil microbial community. PLoS ONE 9:e93445. doi: 10.1371/journal.pone.0093445
Yan, C., Wang, F., Geng, H., Liu, H., Pu, S., Tian, Z., et al. (2020). Integrating high-throughput sequencing and metagenome analysis to reveal the characteristic and resistance mechanism of microbial community in metal contaminated sediments. Sci. Total Environ. 707:106116. doi: 10.1016/j.scitotenv.2019.136116
Yan, X., Jin, W., Wu, G., Jiang, W., Yang, Z., Ji, J., et al. (2018). Hydrolase CehA and monooxygenase CfdC are responsible for carbofuran degradation in Sphingomonas strain CDS-1. Appl. Environ. Microbiol. 84, 1–15. doi: 10.1128/AEM.00805-18
Yang, J., Feng, Y., Zhan, H., Liu, J., Yang, F., Zhang, K., et al. (2018). Characterization of a pyrethroid-degrading Pseudomonas fulva strain P31 and biochemical degradation pathway of D -phenothrin. Front. Microbiol . 9:1003. doi: 10.3389/fmicb.2018.01003
Yang, T., Ren, L., Jia, Y., fan, S., Wang, J., Nahurira, R., et al. (2018). Biodegradation of di-(2-ethylhexyl) phthalate by Rhodococcus ruber YC-YT1 in contaminated water and soil. Int. J. Environ. Res. Public Health. 15:964. doi: 10.3390/ijerph15050964
Yang, Y. (2013). Omics breakthroughs for environmental microbiology. Microbiol. China 40, 18–33.
Yates, J. R., Ruse, C. L., and Nakorchevsky, A. (2009). Proteomics by mass spectrometry: approaches, advances and applications. Ann. Rev. Biomed. Eng . 11, 49–79. doi: 10.1146/annurev-bioeng-061008-124934
Yu, K., Yi, S., Li, B., Guo, F., Peng, X., Wang, Z., et al. (2019). An integrated meta-omics approach reveals substrates involved in synergistic interactions in a bisphenol A (BPA)-degrading microbial community. Microbiome 7:16. doi: 10.1186/s40168-019-0634-5
Yu, Y., Yin, H., Peng, H., Lu, G., and Dang, Z. (2019). Proteomic mechanism of decabromodiphenyl ether (BDE-209) biodegradation by Microbacterium Y2 and its potential in remediation of BDE-209 contaminated water-sediment system. J. Hazard. Mater. 387:121708. doi: 10.1016/j.jhazmat.2019.121708
Zafra, G., Taylor, T. D., Absalon, A. E., and Cortes-Espinosa, D. V. (2016). Comparative metagenomic analysis of PAH degradation in soil by a mixed microbial consortium. J. Hazard. Mater. 318, 702–710. doi: 10.1016/j.jhazmat.2016.07.060
Zepeda, A. E., de Leon, A. V. P., and Flores, A. S. (2015). The road to metagenomics: from microbiology to DNA sequencing technologies and bioinformatics. Front. Genet. 6:648. doi: 10.3389/fgene.2015.00348
Zhan, H., Huang, Y., Lin, Z., Bhatt, P., and Chen, S. (2020). New insights into the microbial degradation and catalytic mechanism of synthetic pyrethroids. Environ. Res. 182:109138. doi: 10.1016/j.envres.2020.109138
Zhan, H., Wang, H., Liao, L., Feng, Y., Fan, X., Zhang, L., et al. (2018). Kinetics and novel degradation pathway of cypermethrin in Acinetobacter baumannii ZH-14. Front. Microbiol. 9:98. doi: 10.3389/fmicb.2018.00098
Zhang, R., Xu, X., Chen, W., and Huang, Q. (2016). Genetically engineered Pseudomonas putida X3 strain and its potential ability to bioremediate soil microcosms contaminated with methyl parathion and cadmium. Appl. Microbiol. Biotechnol. 100, 1987–1997. doi: 10.1007/s00253-015-7099-7
Zhang, W., Lin, Z., Pang, S., Bhatt, P., and Chen, S. (2020). Insights into the biodegradation of lindane (γ-hexacholocyclohexane) using a microbial system. Front. Microbiol. 11:522. doi: 10.3389/fmicb.2020.00522
Zhang, W., Pang, S., Lin, Z., Mishra, S., Bhatt, P., and Chen, S. (2021). Biotransformation of perfluoroalkyl acid precursors from various environmental systems: advances and perspectives. Environ. Pollut. 268:115908. doi: 10.1016/j.envpol.2020.115908
Zhao, Q., Yue, S., Bilal, M., Hu, H., Wang, W., and Zhang, X. (2017). Comparative genomic analysis of 26 Sphingomonas and Sphingobium strains: dissemination of bioremediation capabilities, biodegradation potential and horizontal gene transfer. Sci. Total Environ. 609, 1238–1247. doi: 10.1016/j.scitotenv.2017.07.249
Zhao, X., Ma, F., Feng, C., Bai, S., Yang, J., and Wang, L. (2017). Complete genome sequence of Arthrobacter sp. ZXY-2 associated with effective atrazine degradation and salt adaptation. J. Biotechnol. 248, 43–47. doi: 10.1016/j.jbiotec.2017.03.010
Zhong, Q., Tian, J., Wang, B., and Wang, L. (2016). PMA based real-time fluoresecent LAMP for the detection of Vibrio parahaemolyticus in viable but nonculturable state. Food Control 63, 230–238. doi: 10.1016/j.foodcont.2015.11.043
Zhou, J., He, Z., and Yang, Y. (2015). High-throughput metagenomic technologies for complex microbial community analysis: open and closed formats. mBio 6:e02288-14. doi: 10.1128/mBio.02288-14
Zhu, F., Doyle, E., Zhu, C., Zhou, D., Gu, C., and Gao, J. (2020). Metagenomic analysis exploring microbial assemblages and functional genes potentially involved in di (2-ethylhexyl) phthalate degradation in soil. Sci. Total Environ. 715:137037. doi: 10.1016/j.scitotenv.2020.137037
Zhu, Y., Boye, A., Body-Malape, M., and Herkovits, J. (2017). The toxic effects of xenobiotics on the health of humans and animals. BioMed Res. Int. 17:4627872. doi: 10.1155/2017/4627872
Zhu, Y., Klompe, S. E., Vlot, M., van der Oost, J., and Staals, R. H. (2018). Shooting the messenger: RNA-targetting CRISPR-Cas systems. Biosci. Rep. 38:BSR20170788. doi: 10.1042/BSR20170788
Zierer, J., Jackson, M. A., Kastenmuller, G., Mangino, M., Long, T., Telenti, A., et al. (2018). The fecal metabolome as a functional readout of the gut microbiome. Nat. Genet. 50, 790–795. doi: 10.1038/s41588-018-0135-7
Keywords: bioremediation, microorganisms, xenobiotics, omics, bioinformatics
Citation: Mishra S, Lin Z, Pang S, Zhang W, Bhatt P and Chen S (2021) Recent Advanced Technologies for the Characterization of Xenobiotic-Degrading Microorganisms and Microbial Communities. Front. Bioeng. Biotechnol. 9:632059. doi: 10.3389/fbioe.2021.632059
Received: 22 November 2020; Accepted: 11 January 2021; Published: 10 February 2021.
Reviewed by:
Copyright © 2021 Mishra, Lin, Pang, Zhang, Bhatt and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY) . The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shaohua Chen, shchen@scau.edu.cn
This article is part of the Research Topic
Advanced Bioremediation Technologies and Processes for the Treatment of Synthetic Organic Compounds (SOCs)
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Review Article
- Published: 14 March 2016
The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism
- Peter Spanogiannopoulos 1 ,
- Elizabeth N. Bess 1 ,
- Rachel N. Carmody 1 &
- Peter J. Turnbaugh 1
Nature Reviews Microbiology volume 14 , pages 273–287 ( 2016 ) Cite this article
17k Accesses
404 Citations
144 Altmetric
Metrics details
- Antimicrobials
- Metagenomics
The gut microbiome is a neglected component of the first-pass metabolism of xenobiotics before reaching the general circulation.
Direct microbial metabolism of xenobiotics and their metabolites often involves reduction or hydrolysis, but most of the enzymes responsible for these reactions remain unknown.
Microbial metabolism influences both efficacy and toxicity, producing bioactive compounds, inactive metabolites and toxins.
Relevant host–microbial interactions include the expression of host genes that are involved in drug transport and metabolism, the interference with host enzymatic activity and the modulation of immune responses.
The translational implications of these studies include the development of novel co-therapies and the identification of new biomarkers and drugs.
Although the importance of human genetic polymorphisms in therapeutic outcomes is well established, the role of our 'second genome' (the microbiome) has been largely overlooked. In this Review, we highlight recent studies that have shed light on the mechanisms that link the human gut microbiome to the efficacy and toxicity of xenobiotics, including drugs, dietary compounds and environmental toxins. Continued progress in this area could enable more precise tools for predicting patient responses and for the development of a new generation of therapeutics based on, or targeted at, the gut microbiome. Indeed, the admirable goal of precision medicine may require us to first understand the microbial pharmacists within.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
195,33 € per year
only 16,28 € per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
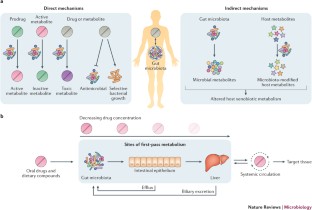
Similar content being viewed by others

Mapping human microbiome drug metabolism by gut bacteria and their genes

Drug-microbiota interactions: an emerging priority for precision medicine

Multimodal interactions of drugs, natural compounds and pollutants with the gut microbiota
Faith, J. J. et al. The long-term stability of the human gut microbiota. Science 341 , 1237439 (2013).
PubMed PubMed Central Google Scholar
Kuczynski, J. et al. Experimental and analytical tools for studying the human microbiome. Nat. Rev. Genet. 13 , 47–58 (2012).
CAS Google Scholar
Maurice, C. F., Haiser, H. J. & Turnbaugh, P. J. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 152 , 39–50 (2013). This study is the first to develop methods to define the metabolically active set of gut bacteria and demonstrate that xenobiotics shape the structure and physiology of these bacteria.
CAS PubMed PubMed Central Google Scholar
Maurice, C. F. & Turnbaugh, P. J. Quantifying the metabolic activities of human-associated microbial communities across multiple ecological scales. FEMS Microbiol. Rev. 37 , 830–848 (2013).
CAS PubMed Google Scholar
O'Hara, A. M. & Shanahan, F. The gut flora as a forgotten organ. EMBO Rep. 7 , 688–693 (2006).
Qin, J. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464 , 59–65 (2010).
Eckburg, P. B. et al. Diversity of the human intestinal microbial flora. Science 308 , 1635–1638 (2005).
David, L. A. et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505 , 559–563 (2014).
Yatsunenko, T. et al. Human gut microbiome viewed across age and geography. Nature 486 , 222–227 (2012).
Goodrich, J. K. et al. Human genetics shape the gut microbiome. Cell 159 , 789–799 (2014).
Thaiss, C. A. et al. Transkingdom control of microbiota diurnal oscillations promotes metabolic homeostasis. Cell 159 , 514–529 (2014).
Diaz Heijtz, R. et al. Normal gut microbiota modulates brain development and behavior. Proc. Natl Acad. Sci. USA 108 , 3047–3052 (2011).
PubMed Google Scholar
Foster, J. A. & McVey Neufeld, K. A. Gut–brain axis: how the microbiome influences anxiety and depression. Trends Neurosci. 36 , 305–312 (2013).
Hsiao, E. Y. et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155 , 1451–1463 (2013).
Arthur, J. C. et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338 , 120–123 (2012).
Kostic, A. D. et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 14 , 207–215 (2013).
Fierer, N. et al. Forensic identification using skin bacterial communities. Proc. Natl Acad. Sci. USA 107 , 6477–6481 (2010).
Franzosa, E. A. et al. Identifying personal microbiomes using metagenomic codes. Proc. Natl Acad. Sci. USA 112 , E2930–E2938 (2015).
Liou, A. P. et al. Conserved shifts in the gut microbiota due to gastric bypass reduce host weight and adiposity. Sci. Transl. Med. 5 , 178ra141 (2013).
Google Scholar
Roopchand, D. E. et al. Dietary polyphenols promote growth of the gut bacterium Akkermansia muciniphila and attenuate high-fat diet-induced metabolic syndrome. Diabetes 64 , 2847–2858 (2015). This study suggests that the beneficial effects of dietary polyphenols may be mediated by the gut microbiome.
Fuller, A. T. Is p -aminobenzenesulphonamide the active agent in protonsil therapy? Lancet 229 , 194–198 (1937).
Colebrook, L., Buttle, G. A. H. & O'Meara, R. A. Q. The mode of action of p -aminobenzene sulphonamide and prontosil in hemolytic streptococcal infections. Lancet 228 , 1323–1326 (1936).
Radomski, J. L. & Mellinger, T. J. The absorption, fate and excretion in rats of the water-soluble azo dyes, FD&C Red No. 2, FD&C Red No. 4, and FD&C Yellow No. 6. J. Pharmacol. Exp. Ther. 136 , 259–266 (1962).
Klotz, U., Maier, K., Fischer, C. & Heinkel, K. Therapeutic efficacy of sulfasalazine and its metabolites in patients with ulcerative colitis and Crohn's disease. N. Engl. J. Med. 303 , 1499–1502 (1980).
Plosker, G. L. & Croom, K. F. Sulfasalazine: a review of its use in the management of rheumatoid arthritis. Drugs 65 , 1825–1849 (2005).
Rocco, T. P. & Fang, J. C. in Goodman & Gilman's The Pharmacological Basis of Therapeutics (eds Brunton, L. L., Lazo, J. S. & Parker, K. L.) (McGraw-Hill, 2011).
Grundmann, O. The gut microbiome and pre-systemic metabolism: current state and evolving research. J. Drug Metab. Toxicol. 1 , 1–7 (2010).
Haiser, H. J. & Turnbaugh, P. J. Developing a metagenomic view of xenobiotic metabolism. Pharmacol. Res. 69 , 21–31 (2013).
Li, H. & Jia, W. Cometabolism of microbes and host: implications for drug metabolism and drug-induced toxicity. Clin. Pharmacol. Ther. 94 , 574–581 (2013).
Nicholson, J. K., Holmes, E. & Wilson, I. D. Gut microorganisms, mammalian metabolism and personalized health care. Nat. Rev. Microbiol. 3 , 431–438 (2005).
Saad, R., Rizkallah, M. R. & Aziz, R. K. Gut pharmacomicrobiomics: the tip of an iceberg of complex interactions between drugs and gut-associated microbes. Gut Pathog. 4 , 16 (2012).
Tralau, T., Sowada, J. & Luch, A. Insights on the human microbiome and its xenobiotic metabolism: what is known about its effects on human physiology? Expert Opin. Drug Metab. Toxicol. 11 , 411–425 (2015).
Pond, S. M. & Tozer, T. N. First-pass elimination. Basic concepts and clinical consequences. Clin. Pharmacokinet. 9 , 1–25 (1984).
Backhed, F., Ley, R. E., Sonnenburg, J. L., Peterson, D. A. & Gordon, J. I. Host–bacterial mutualism in the human intestine. Science 307 , 1915–1920 (2005).
Arkhipova, O. V. & Akimenko, V. K. Unsaturated organic acids as terminal electron acceptors for reductase chains of anaerobic bacteria. Microbiology 76 , 725–737 (2005).
Novel, G., Didier-Fichet, M. L. & Stoeber, F. Inducibility of β-glucuronidase in wild-type and hexuronate-negative mutants of Escherichia coli K-12. J. Bacteriol. 120 , 89–95 (1974).
Haiser, H. J., Seim, K. L., Balskus, E. P. & Turnbaugh, P. J. Mechanistic insight into digoxin inactivation by Eggerthella lenta augments our understanding of its pharmacokinetics. Gut Microbes 5 , 233–238 (2014).
de Groot, M. J. Designing better drugs: predicting cytochrome P450 metabolism. Drug Discov. Today 11 , 601–606 (2006).
Zanger, U. M. & Schwab, M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 138 , 103–141 (2013).
Bachrach, W. H. Sulfasalazine: I. An historical perspective. Am. J. Gastroenterol. 83 , 487–496 (1988).
Svartz, N. Sulfasalazine: II. Some notes on the discovery and development of salazopyrin. Am. J. Gastroenterol. 83 , 497–503 (1988).
Peppercorn, M. A. & Goldman, P. The role of intestinal bacteria in the metabolism of salicylazosulfapyridine. J. Pharmacol. Exp. Ther. 181 , 555–562 (1972).
Chen, H., Wang, R. F. & Cerniglia, C. E. Molecular cloning, overexpression, purification, and characterization of an aerobic FMN-dependent azoreductase from Enterococcus faecalis . Protein Expr. Purif. 34 , 302–310 (2004).
Morrison, J. M., Wright, C. M. & John, G. H. Identification, isolation and characterization of a novel azoreductase from Clostridium perfringens . Anaerobe 18 , 229–234 (2012).
Sousa, T. et al. On the colonic bacterial metabolism of azo-bonded prodrugs of 5-aminosalicylic acid. J. Pharm. Sci. 103 , 3171–3175 (2014).
Delomenie, C. et al. Identification and functional characterization of arylamine N -acetyltransferases in eubacteria: evidence for highly selective acetylation of 5-aminosalicylic acid. J. Bacteriol. 183 , 3417–3427 (2001).
Carmody, R. N. & Turnbaugh, P. J. Host–microbial interactions in the metabolism of therapeutic and diet-derived xenobiotics. J. Clin. Invest. 124 , 4173–4181 (2014).
Wells, P. G. et al. Glucuronidation and the UDP-glucuronosyltransferases in health and disease. Drug Metab. Dispos. 32 , 281–290 (2004).
Wiseman, L. R. & Markham, A. Irinotecan. A review of its pharmacological properties and clinical efficacy in the management of advanced colorectal cancer. Drugs 52 , 606–623 (1996).
Stein, A., Voigt, W. & Jordan, K. Chemotherapy-induced diarrhea: pathophysiology, frequency and guideline-based management. Ther. Adv. Med. Oncol. 2 , 51–63 (2010).
Mani, S., Boelsterli, U. A. & Redinbo, M. R. Understanding and modulating mammalian–microbial communication for improved human health. Annu. Rev. Pharmacol. Toxicol. 54 , 559–580 (2014).
Rothenberg, M. L. et al. Phase II trial of irinotecan in patients with progressive or rapidly recurrent colorectal cancer. J. Clin. Oncol. 14 , 1128–1135 (1996).
Higuchi, K. et al. Present status and strategy of NSAIDs-induced small bowel injury. J. Gastroenterol. 44 , 879–888 (2009).
Saitta, K. S. et al. Bacterial β-glucuronidase inhibition protects mice against enteropathy induced by indomethacin, ketoprofen or diclofenac: mode of action and pharmacokinetics. Xenobiotica 44 , 28–35 (2014). This study demonstrates that the toxicity associated with NSAIDs can be alleviated by inhibiting bacterial enzyme activity with small-molecule inhibitors.
Beaud, D., Tailliez, P. & Anba-Mondoloni, J. Genetic characterization of the β-glucuronidase enzyme from a human intestinal bacterium, Ruminococcus gnavus . Microbiology 151 , 2323–2330 (2005).
Dabek, M., McCrae, S. I., Stevens, V. J., Duncan, S. H. & Louis, P. Distribution of β-glucosidase and β-glucuronidase activity and of β-glucuronidase gene gus in human colonic bacteria. FEMS Microbiol. Ecol. 66 , 487–495 (2008).
Flores, R. et al. Association of fecal microbial diversity and taxonomy with selected enzymatic functions. PLoS ONE 7 , e39745 (2012).
Roy, D. & Ward, P. Rapid detection of Bifidobacterium dentium by enzymatic hydrolysis of β-glucuronide substrates. J. Food Protect. 55 , 291–295 (1992).
Russell, W. M. & Klaenhammer, T. R. Identification and cloning of gusA , encoding a new β-glucuronidase from Lactobacillus gasseri ADH. Appl. Environ. Microbiol. 67 , 1253–1261 (2001).
Wallace, B. D. et al. Structure and inhibition of microbiome β-glucuronidases essential to the alleviation of cancer drug toxicity. Chem. Biol. 22 , 1238–1249 (2015).
Lindenbaum, J. et al. Inactivation of digoxin by the gut flora: reversal by antibiotic therapy. N. Engl. J. Med. 305 , 789–794 (1981).
Matzuk, M. M., Shlomchik, M. & Shaw, L. M. Making digoxin therapeutic drug monitoring more effective. Ther. Drug Monit. 13 , 215–219 (1991).
Peters, U., Falk, L. C. & Kalman, S. M. Digoxin metabolism in patients. Arch. Intern. Med. 138 , 1074–1076 (1978).
Saha, J. R., Butler, V. P., Neu, H. C. & Lindenbaum, J. Digoxin-inactivating bacteria: identification in human gut flora. Science 220 , 325–327 (1983).
Mathan, V. I., Wiederman, J., Dobkin, J. F. & Lindenbaum, J. Geographic differences in digoxin inactivation, a metabolic activity of the human anaerobic gut flora. Gut 30 , 971–977 (1989).
Rowland, I. R. Factors affecting metabolic activity of the intestinal microflora. Drug Metab. Rev. 19 , 243–261 (1988).
Haiser, H. J. et al. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta . Science 341 , 295–298 (2013). This study is the first to show that the bacterial inactivation of drugs can be predicted with a genetic marker and can be prevented using dietary intervention.
Hooper, L. V. et al. Molecular analysis of commensal host–microbial relationships in the intestine. Science 291 , 881–884 (2001).
Bjorkholm, B. et al. Intestinal microbiota regulate xenobiotic metabolism in the liver. PLoS ONE 4 , e6958 (2009).
Lundin, A. et al. Gut flora, Toll-like receptors and nuclear receptors: a tripartite communication that tunes innate immunity in large intestine. Cell. Microbiol. 10 , 1093–1103 (2008).
Claus, S. P. et al. Colonization-induced host–gut microbial metabolic interaction. mBio 2 , e00271-10 (2011).
Selwyn, F. P., Cui, J. Y. & Klaassen, C. D. RNA-seq quantification of hepatic drug processing genes in germ-free mice. Drug Metab. Dispos. 43 , 1572–1580 (2015).
Claus, S. P. et al. Systemic multicompartmental effects of the gut microbiome on mouse metabolic phenotypes. Mol. Syst. Biol. 4 , 219 (2008).
Wikoff, W. R. et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl Acad. Sci. USA 106 , 3698–3703 (2009). This study demonstrates that the microbiome has a substantial role in the signature and abundance of circulating metabolites in mammalian blood.
Hodgman, M. J. & Garrard, A. R. A review of acetaminophen poisoning. Crit. Care Clin. 28 , 499–516 (2012).
Court, M. H. et al. Interindividual variability in acetaminophen glucuronidation by human liver microsomes: identification of relevant acetaminophen UDP-glucuronosyltransferase isoforms. J. Pharmacol. Exp. Ther. 299 , 998–1006 (2001).
Harrill, A. H. et al. Mouse population-guided resequencing reveals that variants in CD44 contribute to acetaminophen-induced liver injury in humans. Genome Res. 19 , 1507–1515 (2009).
Clayton, T. A., Baker, D., Lindon, J. C., Everett, J. R. & Nicholson, J. K. Pharmacometabonomic identification of a significant host–microbiome metabolic interaction affecting human drug metabolism. Proc. Natl Acad. Sci. USA 106 , 14728–14733 (2009). This study is the first to demonstrate the potential of using levels of microbial metabolites as predictive biomarkers for drug metabolism.
Bone, E., Tamm, A. & Hill, M. The production of urinary phenols by gut bacteria and their possible role in the causation of large bowel cancer. Am. J. Clin. Nutr. 29 , 1448–1454 (1976).
Selmer, T. & Andrei, P. I. p -hydroxyphenylacetate decarboxylase from Clostridium difficile . A novel glycyl radical enzyme catalysing the formation of p -cresol. Eur. J. Biochem. 268 , 1363–1372 (2001).
Gamage, N. et al. Human sulfotransferases and their role in chemical metabolism. Toxicol. Sci. 90 , 5–22 (2006).
Mangravite, L. M., Thorn, C. F. & Krauss, R. M. Clinical implications of pharmacogenomics of statin treatment. Pharmacogenom. J. 6 , 360–374 (2006).
Kaddurah-Daouk, R. et al. Enteric microbiome metabolites correlate with response to simvastatin treatment. PLoS ONE 6 , e25482 (2011).
Mitchell, J. B. et al. A low molecular weight antioxidant decreases weight and lowers tumor incidence. Free Radic. Biol. Med. 34 , 93–102 (2003).
Li, F. et al. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat. Commun. 4 , 2384 (2013).
Jiang, J. et al. Diversity of bile salt hydrolase activities in different lactobacilli toward human bile salts. Ann. Microbiol. 60 , 81–88 (2010).
de Wit, N. J. et al. The role of the small intestine in the development of dietary fat-induced obesity and insulin resistance in C57BL/6J mice. BMC Med. Genom. 1 , 14 (2008).
Sayin, S. I. et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-β-muricholic acid, a naturally occurring FXR antagonist. Cell. Metab. 17 , 225–235 (2013).
Tsuda, T. Regulation of adipocyte function by anthocyanins; possibility of preventing the metabolic syndrome. J. Agr. Food Chem. 56 , 642–646 (2008).
Quesada, H. et al. Grape seed proanthocyanidins correct dyslipidemia associated with a high-fat diet in rats and repress genes controlling lipogenesis and VLDL assembling in liver. Int. J. Obes. (Lond.) 33 , 1007–1012 (2009).
Tsuda, T., Horio, F., Uchida, K., Aoki, H. & Osawa, T. Dietary cyanidin 3- O -β-d-glucoside-rich purple corn color prevents obesity and ameliorates hyperglycemia in mice. J. Nutr. 133 , 2125–2130 (2003).
Baiges, I., Palmfeldt, J., Blade, C., Gregersen, N. & Arola, L. Lipogenesis is decreased by grape seed proanthocyanidins according to liver proteomics of rats fed a high fat diet. Mol. Cell. Proteom. 9 , 1499–1513 (2010).
Cefalu, W. T. et al. Botanicals and the metabolic syndrome. Am. J. Clin. Nutr. 87 , 481S–487S (2008).
Felgines, C. et al. Radiolabelled cyanidin 3- O -glucoside is poorly absorbed in the mouse. Br. J. Nutr. 103 , 1738–1745 (2010).
Abia, R. & Fry, S. C. Degradation and metabolism of 14 C-labelled proanthocyanidins from carob ( Ceratonia siliqua ) pods in the gastrointestinal tract of the rat. J. Sci. Food Agr. 81 , 1156–1165 (2001).
Anhe, F. F. et al. A polyphenol-rich cranberry extract protects from diet-induced obesity, insulin resistance and intestinal inflammation in association with increased Akkermansia spp. population in the gut microbiota of mice. Gut 64 , 872–883 (2015).
Everard, A. et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl Acad. Sci. USA 110 , 9066–9071 (2013).
Santacruz, A. et al. Gut microbiota composition is associated with body weight, weight gain and biochemical parameters in pregnant women. Br. J. Nutr. 104 , 83–92 (2010).
Zhang, H. et al. Human gut microbiota in obesity and after gastric bypass. Proc. Natl Acad. Sci. USA 106 , 2365–2370 (2009).
Everard, A. et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 60 , 2775–2786 (2011).
Shin, N. R. et al. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 63 , 727–735 (2014). This study suggests that the anti-diabetic drug metformin may act, in part, by altering the gut microbiome.
Axling, U. et al. Green tea powder and Lactobacillus plantarum affect gut microbiota, lipid metabolism and inflammation in high-fat fed C57BL/6J mice. Nutr. Metab. (Lond.) 9 , 105 (2012).
Kemperman, R. A. et al. Impact of polyphenols from black tea and red wine/grape juice on a gut model microbiome. Food Res. Int. 53 , 659–669 (2013).
Haslam, E. Natural polyphenols (vegetable tannins) as drugs: possible modes of action. J. Nat. Prod. 59 , 205–215 (1996).
Nunez-Sanchez, M. A. et al. Targeted metabolic profiling of pomegranate polyphenols and urolithins in plasma, urine and colon tissues from colorectal cancer patients. Mol. Nutr. Food. Res. 58 , 1199–1211 (2014).
Garcia-Munoz, C. & Vaillant, F. Metabolic fate of ellagitannins: implications for health, and research perspectives for innovative functional foods. Crit. Rev. Food Sci. Nutr. 54 , 1584–1598 (2014).
Tomas-Barberan, F. A., Garcia-Villalba, R., Gonzalez-Sarrias, A., Selma, M. V. & Espin, J. C. Ellagic acid metabolism by human gut microbiota: consistent observation of three urolithin phenotypes in intervention trials, independent of food source, age, and health status. J. Agr. Food Chem. 62 , 6535–6538 (2014).
Garcia-Villalba, R., Beltran, D., Espin, J. C., Selma, M. V. & Tomas-Barberan, F. A. Time course production of urolithins from ellagic acid by human gut microbiota. J. Agr. Food Chem. 61 , 8797–8806 (2013).
Selma, M. V., Beltran, D., Garcia-Villalba, R., Espin, J. C. & Tomas-Barberan, F. A. Description of urolithin production capacity from ellagic acid of two human intestinal Gordonibacter species. Food Funct. 5 , 1779–1784 (2014).
Adlercreutz, H. Lignans and human health. Crit. Rev. Clin. Lab Sci. 44 , 483–525 (2007).
Lampe, J. W. Is equol the key to the efficacy of soy foods? Am. J. Clin. Nutr. 89 , 1664S–1667S (2009).
Patisaul, H. B. & Jefferson, W. The pros and cons of phytoestrogens. Front. Neuroendocrinol. 31 , 400–419 (2010).
Setchell, K. D. & Clerici, C. Equol: history, chemistry, and formation. J. Nutr. 140 , 1355S–1362S (2010).
Wu, A. H., Yu, M. C., Tseng, C. C. & Pike, M. C. Epidemiology of soy exposures and breast cancer risk. Br. J. Cancer 98 , 9–14 (2008).
Setchell, K. D., Brown, N. M. & Lydeking-Olsen, E. The clinical importance of the metabolite equol-a clue to the effectiveness of soy and its isoflavones. J. Nutr. 132 , 3577–3584 (2002).
Duncan, A. M., Merz-Demlow, B. E., Xu, X., Phipps, W. R. & Kurzer, M. S. Premenopausal equol excretors show plasma hormone profiles associated with lowered risk of breast cancer. Cancer Epidemiol. Biomarkers Prev. 9 , 581–586 (2000).
Virk-Baker, M. K., Barnes, S., Krontiras, H. & Nagy, T. R. S-(−)equol producing status not associated with breast cancer risk among low isoflavone-consuming US postmenopausal women undergoing a physician-recommended breast biopsy. Nutr. Res. 34 , 116–125 (2014).
Mazur, W. & Adlercreutz, H. Natural and anthropogenic environmental oestrogens: the scientific basis for risk assessment. Pure Appl. Chem. 70 , 1759–1776 (1998).
Penalvo, J. L., Haajanen, K. M., Botting, N. & Adlercreutz, H. Quantification of lignans in food using isotope dilution gas chromatography/mass spectrometry. J. Agr. Food Chem. 53 , 9342–9347 (2005).
Clavel, T., Borrmann, D., Braune, A., Dore, J. & Blaut, M. Occurrence and activity of human intestinal bacteria involved in the conversion of dietary lignans. Anaerobe 12 , 140–147 (2006).
Mabrok, H. B. et al. Lignan transformation by gut bacteria lowers tumor burden in a gnotobiotic rat model of breast cancer. Carcinogenesis 33 , 203–208 (2012). This study determines that the microbial production of bioactive metabolites from dietary sources mediates anticancer effects for the host.
Kaderlik, K. R. et al. Glucuronidation of N -hydroxy heterocyclic amines by human and rat liver microsomes. Carcinogenesis 15 , 1695–1701 (1994).
Hirayama, K. et al. Effects of human intestinal flora on mutagenicity of and DNA adduct formation from food and environmental mutagens. Carcinogenesis 21 , 2105–2111 (2000).
Kassie, F. et al. Intestinal microflora plays a crucial role in the genotoxicity of the cooked food mutagen 2-amino-3-methylimidazo[4,5- f ]quinoline. Carcinogenesis 22 , 1721–1725 (2001).
Humblot, C. et al. β-glucuronidase in human intestinal microbiota is necessary for the colonic genotoxicity of the food-borne carcinogen 2-amino-3-methylimidazo[4,5- f ]quinoline in rats. Carcinogenesis 28 , 2419–2425 (2007). This study shows that microbial glucuronidation activity in the gut contributes to the carcinogenic effect of heterocyclic amines from charred meat.
Craciun, S. & Balskus, E. P. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc. Natl Acad. Sci. USA 109 , 21307–21312 (2012).
Wang, Z. et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472 , 57–63 (2011). This study demonstrates that the metabolism of dietary lipids by gut bacteria contributes to heart disease.
Tang, W. H. et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 368 , 1575–1584 (2013).
Koeth, R. A. et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 19 , 576–585 (2013).
Drasar, B. S., Renwick, A. G. & Williams, R. T. The role of the gut flora in the metabolism of cyclamate. Biochem. J. 129 , 881–890 (1972).
Legator, M. S., Palmer, K. A., Green, S. & Petersen, K. W. Cytogenetic studies in rats of cyclohexylamine, a metabolite of cyclamate. Science 165 , 1139–1140 (1969).
Tamura, M., Hoshi, C. & Hori, S. Xylitol affects the intestinal microbiota and metabolism of daidzein in adult male mice. Int. J. Mol. Sci. 14 , 23993–24007 (2013).
Suez, J. et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 514 , 181–186 (2014).
Brown, R. J., de Banate, M. A. & Rother, K. I. Artificial sweeteners: a systematic review of metabolic effects in youth. Int. J. Pediatr. Obes. 5 , 305–312 (2010).
Chassaing, B. et al. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 519 , 92–96 (2015). This research shows that dietary supplements can disrupt mucus–bacterial interactions, promoting gut inflammation.
Hau, A. K., Kwan, T. H. & Li, P. K. Melamine toxicity and the kidney. J. Am. Soc. Nephrol. 20 , 245–250 (2009).
Ingelfinger, J. R. Melamine and the global implications of food contamination. N. Engl. J. Med. 359 , 2745–2748 (2008).
Zheng, X. et al. Melamine-induced renal toxicity is mediated by the gut microbiota. Sci. Transl. Med. 5 , 172ra122 (2013). This study attributes the toxicity of dietary contaminants to gut microbial metabolism.
Shelton, D. R., Karns, J. S., McCarty, G. W. & Durham, D. R. Metabolism of melamine by Klebsiella terragena . Appl. Environ. Microbiol. 63 , 2832–2835 (1997).
Jutzi, K., Cook, A. M. & Hutter, R. The degradative pathway of the s -triazine melamine. The steps to ring cleavage. Biochem. J. 208 , 679–684 (1982).
Podschun, R. Isolation of Klebsiella terrigena from human feces: biochemical reactions, capsule types, and antibiotic sensitivity. Zentralbl. Bakteriol. 275 , 73–78 (1991).
Sonnenburg, J. L. & Fischbach, M. A. Community health care: therapeutic opportunities in the human microbiome. Sci. Transl. Med. 3 , 78ps12 (2011).
Ahmad, S. et al. A high throughput assay for discovery of bacterial β-glucuronidase inhibitors. Curr. Chem. Genom. 5 , 13–20 (2011).
Ahmad, S., Hughes, M. A., Yeh, L. A. & Scott, J. E. Potential repurposing of known drugs as potent bacterial β-glucuronidase inhibitors. J. Biomol. Screen 17 , 957–965 (2012).
Roberts, A. B., Wallace, B. D., Venkatesh, M. K., Mani, S. & Redinbo, M. R. Molecular insights into microbial β-glucuronidase inhibition to abrogate CPT-11 toxicity. Mol. Pharmacol. 84 , 208–217 (2013).
Wallace, B. D. et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 330 , 831–835 (2010). This is the first study to show that the toxicity associated with the microbial metabolism of cancer drugs in the gut can be mitigated using bacterial enzyme-specific small-molecule inhibitors.
LoGuidice, A., Wallace, B. D., Bendel, L., Redinbo, M. R. & Boelsterli, U. A. Pharmacologic targeting of bacterial β-glucuronidase alleviates nonsteroidal anti-inflammatory drug-induced enteropathy in mice. J. Pharmacol. Exp. Ther. 341 , 447–454 (2012).
Carmody, R. N. et al. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 17 , 72–84 (2015).
Lee, J. R. et al. Gut microbiota and tacrolimus dosing in kidney transplantation. PLoS ONE 10 , e0122399 (2015).
Hooper, L. V., Littman, D. R. & Macpherson, A. J. Interactions between the microbiota and the immune system. Science 336 , 1268–1273 (2012).
Viaud, S. et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 342 , 971–976 (2013).
Viaud, S. et al. Cyclophosphamide induces differentiation of TH17 cells in cancer patients. Cancer Res. 71 , 661–665 (2011).
Iida, N. et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 342 , 967–970 (2013).
Ozben, T. Oxidative stress and apoptosis: impact on cancer therapy. J. Pharm. Sci. 96 , 2181–2196 (2007).
Sivan, A. et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350 , 1084–1089 (2015).
Vetizou, M. et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350 , 1079–1084 (2015).
Garrett, W. S. et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131 , 33–45 (2007).
Neurath, M. F. New targets for mucosal healing and therapy in inflammatory bowel diseases. Mucosal Immunol. 7 , 6–19 (2014).
Rooks, M. G. et al. Gut microbiome composition and function in experimental colitis during active disease and treatment-induced remission. ISME J. 8 , 1403–1417 (2014).
Donia, M. S. et al. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell 158 , 1402–1414 (2014). This study reveals the potential of the human microbiome as a rich source of bioactive natural products, including antibiotics.
Rupnik, M., Wilcox, M. H. & Gerding, D. N. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat. Rev. Microbiol. 7 , 526–536 (2009).
Buffie, C. G. et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile . Nature 517 , 205–208 (2015).
Sorg, J. A. & Sonenshein, A. L. Inhibiting the initiation of Clostridium difficile spore germination using analogs of chenodeoxycholic acid, a bile acid. J. Bacteriol. 192 , 4983–4990 (2010).
Wilson, K. H. Efficiency of various bile salt preparations for stimulation of Clostridium difficile spore germination. J. Clin. Microbiol. 18 , 1017–1019 (1983).
Download references
Acknowledgements
The authors apologize to all of those colleagues whose work could not be included in this Review owing to space constraints. The authors also thank the reviewers for their comments and suggestions. This work was supported by the US National Institutes of Health (R01AT008618, R01HL122593 and F32DK101154), the Young Investigator Grant for Probiotics Research, the George Williams Hooper Research Foundation and the University of California San Francisco (UCSF) Department of Microbiology & Immunology. P.J.T. is a Nadia's Gift Foundation Innovator supported, in part, by the Damon Runyon Cancer Research Foundation (DRR-42-16).
Author information
Authors and affiliations.
Department of Microbiology & Immunology, George Williams Hooper Foundation, University of California San Francisco, 513 Parnassus Avenue, San Francisco, 94143, California, USA
Peter Spanogiannopoulos, Elizabeth N. Bess, Rachel N. Carmody & Peter J. Turnbaugh
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Peter J. Turnbaugh .
Ethics declarations
Competing interests.
P.J.T. is on the Scientific Advisory Board for Seres Therapeutics and Whole Biome, has consulted for Pfizer in the past year and has current research support from MedImmune.
Supplementary information
Comprehensive list of pharmaceuticals and dietary compounds subject to gut microbial metabolism (PDF 591 kb)
PowerPoint slides
Powerpoint slide for fig. 1, powerpoint slide for fig. 2, powerpoint slide for fig. 3, powerpoint slide for fig. 4, powerpoint slide for fig. 5.
The combined genetic material and metabolic activities of the microbiota.
The collection of all microorganisms (archaea, bacteria, microscopic fungi, parasites and viruses) found in a given body habitat.
Compounds that are foreign to a biological system. For humans, these include drugs, dietary bioactive compounds, food additives and environmental toxins.
A chemical bond composed of N=N.
The study of how genetic factors influence therapeutic outcomes.
The use of sequencing-based genomic methods to analyse the links between genetics and therapeutic outcomes.
The proportion of an administered compound that reaches systemic circulation and thus has the potential to influence the intended target.
The metabolism of orally ingested compounds before reaching general circulation.
The transfer of xenobiotics and other compounds from the plasma to bile through hepatocytes, which is followed by the release of the compounds into the gut lumen.
The circulation of xenobiotics and endogenous compounds that are absorbed from the intestines, transported to the liver, and then re-enter the intestine through the bile ducts, where they may be reabsorbed or metabolized by the gut microbiota.
A chemical reaction in which the oxidation state of a chemical bond is reduced. For example, a carbon–carbon bond modified to a carbon–hydrogen bond is a reductive transformation.
A chemical reaction in which a chemical bond is cleaved using a water molecule, which acts as the nucleophile.
(CYPs) A family of enzymes that is responsible for the oxidative biotransformation of xenobiotics and other compounds.
Drugs that are administered in an inactive form and become active when metabolized.
A B vitamin that is essential for DNA synthesis, DNA repair and other biological reactions.
Animals devoid of microorganisms.
The colonization of germ-free animals with individual microorganisms or defined microbial communities.
The addition of glucuronic acid to a substrate. Glucuronidation is used as a mechanism of xenobiotic metabolism by the host.
Steroid acids produced by the liver that emulsify fats during digestion.
The remaining compound after the removal of a glycosyl moiety.
The collection of all metabolites found in serum.
The addition of a chemical unit (for example, glucuronic acid or glutathione) to xenobiotics, increasing the solubility and molecular weight of the parent compound and facilitating elimination from the body.
A collection of physiological and biochemical conditions, defined as a combination of high blood pressure, increased blood sugar levels, excess fat and abnormal cholesterol levels. This syndrome increases the risk of heart disease, stroke and diabetes.
An oral medication used to treat type 2 diabetes.
A manual for the preparation and use of medicinal drugs. The name is derived from the Greek words pharmakon (drug) and - poios (making).
Rights and permissions
Reprints and permissions
About this article
Cite this article.
Spanogiannopoulos, P., Bess, E., Carmody, R. et al. The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nat Rev Microbiol 14 , 273–287 (2016). https://doi.org/10.1038/nrmicro.2016.17
Download citation
Published : 14 March 2016
Issue Date : May 2016
DOI : https://doi.org/10.1038/nrmicro.2016.17
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
This article is cited by
Gut microbiota, circulating cytokines and dementia: a mendelian randomization study.
- Wen-Zhu Chen
- Li-Jian Chen
Journal of Neuroinflammation (2024)
Gut microbiome modulates tacrolimus pharmacokinetics through the transcriptional regulation of ABCB1
- Alexandra L. Degraeve
- Vincent Haufroid
- Laure B. Bindels
Microbiome (2023)
Genome-scale metabolic reconstruction of 7,302 human microorganisms for personalized medicine
- Almut Heinken
- Johannes Hertel
- Ines Thiele
Nature Biotechnology (2023)
Sexual Dimorphic Interplays Between Gut Microbiota and Antihypertensive Drugs
- Pritam Bardhan
Current Hypertension Reports (2023)
Altered gastrointestinal tract structure and microbiome following cerebral malaria infection
- Sarah A. Knowler
- Anya Shindler
- Ashley E. Franks
Parasitology Research (2023)
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing: Microbiology newsletter — what matters in microbiology research, free to your inbox weekly.

Recent Advanced Technologies for the Characterization of Xenobiotic-Degrading Microorganisms and Microbial Communities
Affiliations.
- 1 State Key Laboratory for Conservation and Utilization of Subtropical Agro-Bioresources, Guangdong Province Key Laboratory of Microbial Signals and Disease Control, Integrative Microbiology Research Centre, South China Agricultural University, Guangzhou, China.
- 2 Guangdong Laboratory for Lingnan Modern Agriculture, Guangzhou, China.
- PMID: 33644024
- PMCID: PMC7902726
- DOI: 10.3389/fbioe.2021.632059
Global environmental contamination with a complex mixture of xenobiotics has become a major environmental issue worldwide. Many xenobiotic compounds severely impact the environment due to their high toxicity, prolonged persistence, and limited biodegradability. Microbial-assisted degradation of xenobiotic compounds is considered to be the most effective and beneficial approach. Microorganisms have remarkable catabolic potential, with genes, enzymes, and degradation pathways implicated in the process of biodegradation. A number of microbes, including Alcaligenes, Cellulosimicrobium, Microbacterium, Micrococcus, Methanospirillum, Aeromonas, Sphingobium, Flavobacterium, Rhodococcus, Aspergillus, Penecillium, Trichoderma, Streptomyces, Rhodotorula, Candida , and Aureobasidium , have been isolated and characterized, and have shown exceptional biodegradation potential for a variety of xenobiotic contaminants from soil/water environments. Microorganisms potentially utilize xenobiotic contaminants as carbon or nitrogen sources to sustain their growth and metabolic activities. Diverse microbial populations survive in harsh contaminated environments, exhibiting a significant biodegradation potential to degrade and transform pollutants. However, the study of such microbial populations requires a more advanced and multifaceted approach. Currently, multiple advanced approaches, including metagenomics, proteomics, transcriptomics, and metabolomics, are successfully employed for the characterization of pollutant-degrading microorganisms, their metabolic machinery, novel proteins, and catabolic genes involved in the degradation process. These technologies are highly sophisticated, and efficient for obtaining information about the genetic diversity and community structures of microorganisms. Advanced molecular technologies used for the characterization of complex microbial communities give an in-depth understanding of their structural and functional aspects, and help to resolve issues related to the biodegradation potential of microorganisms. This review article discusses the biodegradation potential of microorganisms and provides insights into recent advances and omics approaches employed for the specific characterization of xenobiotic-degrading microorganisms from contaminated environments.
Keywords: bioinformatics; bioremediation; microorganisms; omics; xenobiotics.
Copyright © 2021 Mishra, Lin, Pang, Zhang, Bhatt and Chen.
Publication types
- Systematic Review
Cookies on GOV.UK
We use some essential cookies to make this website work.
We’d like to set additional cookies to understand how you use GOV.UK, remember your settings and improve government services.
We also use cookies set by other sites to help us deliver content from their services.
You have accepted additional cookies. You can change your cookie settings at any time.
You have rejected additional cookies. You can change your cookie settings at any time.
- Health and social care
- Medicines, medical devices
- Clinical trials and investigations
VDEC is supporting a GBS vaccine to prevent newborn deaths
Antimicrobial resistance to Group B Streptococcus (GBS) antibiotics is growing. This puts newborns at risk. VDEC is supporting a new vaccine to cut antibiotic use

The vaccine development and evaluation centre ( VDEC ) facilitates the development and evaluation of new vaccines and therapeutics.
VDEC ’s work includes vaccine discovery and surveillance . The Vaccine Assay and Immune Response team within VDEC provides world class scientific expertise and regulated facilities to enable the classification of diseases, pathogens and their mechanisms of action. They also provide continued national surveillance after the release of vaccines.
They have recently been supporting the creation of a maternal vaccine for Group B Streptococcus ( GBS ).
Work with VDEC
We work with industry, academia and government. Contact UKHSA today to see how we can help you.
Get in touch
GBS is the leading cause of vaccine-preventable infections in newborns in the developed world, and a significant cause of newborn infections and stillbirths worldwide. As such, GBS is a leading driver of antibiotic use in neonatal settings, and antimicrobial resistance is increasing.
To reduce early-onset disease (usually within the first 48 hours of life) many countries have introduced screening for GBS in pregnancy. Culture positive mothers are then given intravenous antibiotics in childbirth to protect both the mother and the infant. This has dramatically increased the use of antibiotics in childbirth. In some countries over 50% of childbirths now involve intravenous antibiotic use.
A maternal vaccine for GBS which protects infants from both early and late onset disease (for which intrapartum antibiotics have no effect) will have a positive impact on infant mortality and morbidity. It will also lead to a sharp reduction in the use of antibiotics in neonatal units worldwide.
Reducing antibiotic use in newborns will allow the development of appropriate commensal gut bacteria. This enables them to digest milk and reduces the risk of antimicrobial resistance.
The UKHSA Pathogen Immunology Group at Porton Down had previously been part of the GASTON consortium. This is an international consortium to develop standardised correlates of protection to allow the evaluation of new GBS vaccines and enable licensure of the leading candidates.
UKHSA Porton successfully developed a ‘Gold Standard’ opsonophagocytic killing assay ( OPKA ) and led a global interlaboratory study of the OPKA in public health, academic and industry labs. The assay was shown to be highly sensitive and reproducible in different laboratories, and standard GBS test strains have been selected by UKHSA and made available worldwide.
What VDEC offers
In 2022 to 2023, follow-on funding was awarded by the Bill and Melinda Gates Foundation ( BMGF ) to develop a higher-throughput version of the OPKA suitable for Phase II/III clinical trials and to provide international reference serum samples with defined units of opsonophagocytic activity. The high throughput OPKA is required as the current assay method is very labour-intensive which limits the number of sera that can be analysed in large vaccine studies.
Expected outcome
Licensure of a GBS vaccine will:
- prevent the deaths of tens of thousands of newborns worldwide
- reduce the need for antibiotic treatment in disease cases
- reduce the need for intrapartum antibiotics during childbirth
Future work
As part of this project, the team has also developed an OPKA for GBS serotype VII, which will be used in a follow-up PATH -funded Phase I/II clinical study of a novel 6-valent GBS vaccine currently being developed.
Is this page useful?
- Yes this page is useful
- No this page is not useful
Help us improve GOV.UK
Don’t include personal or financial information like your National Insurance number or credit card details.
To help us improve GOV.UK, we’d like to know more about your visit today. Please fill in this survey (opens in a new tab) .
Study links talc use to ovarian cancer — a potential boon for thousands suing J&J

New research published this week lends credence to the more than 50,000 lawsuits against Johnson & Johnson that allege its talc-based baby powder caused ovarian cancer.
The analysis , released Wednesday in the Journal of Clinical Oncology, found that applying talc powder to the genitals was associated with ovarian cancer — and that the association was greater for people who used the powder frequently or for long periods of time.
The researchers are from the National Institutes of Health, and their findings were based on data from the Sister Study, which enrolled more than 50,000 women in the U.S. from 2003 to 2009. The participants joined when they were between 35 and 74 years old, and each had a sister who’d been diagnosed with breast cancer, which might put them at increased risk for breast or ovarian cancer.
Lawsuits related to J&J’s talc-based baby powder date back to 1999, when a woman alleged that a lifetime of using it led to her mesothelioma, a rare cancer usually caused by exposure to asbestos — a known carcinogen. In 2009, another woman sued the company, alleging that its talc-based products caused her ovarian cancer. Since then, many thousands of others have filed claims over cases of ovarian cancer or mesothelioma that they say were caused by asbestos in J&J baby powder.
J&J has stood by the safety of its talc products and denies that they ever contained asbestos. The company has also argued that studies have not demonstrated a convincing link between ovarian cancer and talc-based products.
The new research could undermine that line of reasoning as the legal battles continue. Most of the lawsuits against J&J have been consolidated into a single federal case in New Jersey, with trial scheduled for December.
“This study is quite timely. We feel like it completely affirms and confirms the position taken by plaintiffs’ experts,” said Leigh O’Dell, a principal at Beasley Allen Law Firm. O’Dell is the co-lead counsel for the plaintiffs’ steering committee, a group of attorneys appointed to act on behalf of the many people with pending cases against J&J.
However, Erik Haas, J&J’s worldwide vice president of litigation, said the new analysis doesn’t establish causality or implicate a specific cancer-inducing agent.
“This study does not change the overwhelming evidence that talcum powder does not cause ovarian cancer,” he said.
Earlier this month, J&J proposed a payment of around $6.48 billion to resolve the lawsuits, but the deal would involve moving the cases to bankruptcy court and require 75% of claimants to vote in favor.
J&J has tried and failed twice to resolve talc lawsuits in bankruptcy court . The company created a subsidiary in 2021 that could assume liability for talc-related legal claims — a legal maneuver known as a Texas two-step. But thus far, courts have dismissed the bankruptcy filings on the grounds that the subsidiary is not in financial distress.

O’Dell said her team “would like to see these women offered a reasonable and fair resolution outside of bankruptcy.”
“Any effort to file another bankruptcy, we believe, is just yet another abuse of the bankruptcy system,” she said.
The potential harms of talc products
The new study asked women how often they used talc powder on their genitals from ages 10 to 13 and during the year before they enrolled in the study. NIH researchers followed up with surveys from 2017 to 2019 that asked women about their lifetime use of talc powder.
Based on the responses, the researchers estimated that up to 56% of the women used talc powder on their genitals at some point. These women were more likely to be Black, less educated and live in the South compared with people who didn’t use talc powder.
The analysis can’t prove that talc causes ovarian cancer, nor does it identify a brand or chemical driving the association. Dale Sandler, one of the study’s authors and the chief of the epidemiology branch at the National Institute of Environmental Health Sciences, said there probably isn’t a way to prove causality in human studies.
“You can’t do a clinical trial and randomize people to ‘powder’ and ‘no powder.’ So we’re going to need to look to other types of research,” she said.
At the very least, the findings should prompt women to rethink their use of talc products, said Katie O’Brien, the lead author of the analysis and an epidemiologist at the National Institute of Environmental Health Sciences.
“We’re not aware of any medically necessary reasons why someone would need to use talcum,” she said.
Current formulations of J&J baby powder use cornstarch, not talc. The company pulled the talc-based versions from the North American market in 2020, citing waning demand and “misinformation around the safety of the product,” and discontinued the product internationally last year.
Talc and asbestos are found in close proximity in nature, so some raw talc collected via mining may be contaminated with asbestos , according to the Food and Drug Administration.
A 2018 Reuters investigation suggested that J&J knew some of its baby powder was contaminated with small amounts of asbestos as early as the 1970s. But J&J denies asbestos was ever present in its products.
O’Brien said asbestos might not be the only reason for an association between talc and cancer. Some talc products may also contain phthalates — chemicals that disrupt hormones in the body and have been linked to ovarian cancer . Plus, talc itself can be abrasive, she added, so it may cause inflammation in the areas where it’s applied. Inflammation is independently associated with the development of cancer.
A debate over the science
Debates over the research linking talc and ovarian cancer will almost certainly be a focus of upcoming litigation in the J&J case.
The New Jersey federal court ruled in March that the company can contest findings that link ovarian cancer to talc.
To support its position, J&J has pointed to research that O’Brien and Sandler published in 2020 , which did not find a statistically significant association between ovarian cancer and the use of talc powder.
But O’Brien said that older study may not have been set up to detect small changes in risk because it did not ask women about their lifetime use or factor in the chance that people might misremember their past habits. Sandler said the new study accounts for those two variables.
“This newer analysis sort of tips the balance by accounting for all these possible ways that reporting could have been incomplete in the prior literature,” she said.
How talc may have played into body shame
J&J started selling talc-based baby powder in 1894.
Although many women have used it to keep their genitals dry, there’s no need to use powder to get rid of moisture in that area, said Alexandra Scranton, director of science and research at Women’s Voices for the Earth, a nonprofit that aims to eliminate chemicals that negatively affect women’s health.
“Moisture in this part of the body is a very healthy thing,” Scranton said. “This part of the body is covered in mucous membranes. It’s supposed to be moist.”
According to O’Brien’s research, some women in the 2000s — often those in their 20s and 30s — also used talc powder on their genitals to feel clean and reduce odor. That application isn’t advised by health experts, either, since the vagina is self-cleaning and good bacteria inside of it naturally produce a slight odor.
Companies like J&J were “basically creating and promoting this myth that this part of your body — your genitals, your vagina — are inherently dirty and that they’re inherently odorous, and therefore inherently shameful,” Scranton said.
J&J said it disagrees with that characterization.
Some women continue to use baby powder on their genitals or have adopted new products like vaginal washes or scented body deodorants.
“It’s so ingrained and so part of the way they take care of their bodies that they can’t imagine not doing it,” Scranton said. “They’ve got their mom’s voice in their head: ‘This is what you do to be a respectable woman.’”
Aria Bendix is the breaking health reporter for NBC News Digital.
- SUGGESTED TOPICS
- The Magazine
- Newsletters
- Managing Yourself
- Managing Teams
- Work-life Balance
- The Big Idea
- Data & Visuals
- Reading Lists
- Case Selections
- HBR Learning
- Topic Feeds
- Account Settings
- Email Preferences
How One Company Added Carbon Estimates to Its Customer Invoices
- Robert S. Kaplan
- Timmy Melotte

A four-step playbook to help businesses increase transparency and reduce emissions.
Soprema is an international building materials supplier, producing millions of square meters of waterproofing, insulating, and roofing products each year. In 2022, Pierre-Etienne Bindschedler, the company’s president and third-generation owner, committed to reporting the carbon footprint of each product on every customer invoice, and to help customers reduce the embedded GHG emissions in the products they purchased. Paper co-author Melotte, an experienced operations director, was selected to lead a pilot project to measure and subsequently lower the carbon embedded in its products. Melotte decided to follow the E-Liability Pilot Playbook, which divides a pilot project into four stages: Project Design, Data Collection; Data Analysis, and Action. This article describes how the pilot, which focused on the company’s bitumen waterproofing systems, unfolded at Soprema. The company estimates a potential carbon footprint reduction of 34% from the project.
In 2022, Pierre-Etienne Bindschedler, the president and third-generation owner of Soprema, set a goal to develop sustainable solutions for customers. Soprema is a multi-product, family-owned business in the middle of the building materials value chain and produces millions of square meters of waterproofing, insulating, and roofing products each year. Bindschedler wanted to report the carbon footprint of each product on every customer invoice, and to help customers reduce the embedded GHG emissions in the products they purchased.
- Robert S. Kaplan is a senior fellow and the Marvin Bower Professor of Leadership Development emeritus at Harvard Business School. He coauthored the McKinsey Award–winning HBR article “ Accounting for Climate Change ” (November–December 2021).
- TM Timmy Melotte is an Operational Excellence Director for Soprema International, a building materials supplier based in Limburg, Belgium
Partner Center

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Microorganisms
Special Issue: Microbial Degradation of Xenobiotics
Xenobiotics are released into the environment by human activities, and they often cause problems such as environmental pollution, since most such compounds cannot be readily degraded, and have harmful effects on human beings and the natural ecosystem. However, some microorganisms that degrade man-made xenobiotics have been isolated. Most of these aerobic xenobiotics-degrading bacterial strains use xenobiotics as their sole source of carbon and energy, and thus they are excellent models for studying the adaptation and evolution of bacteria in the environment.
Recent genome analyses of bacterial strains that degrade xenobiotics have strongly suggested that they indeed emerged relatively recently by gathering genes for the degradation of xenobiotics, and mobile genetic elements played important roles in the recruitment of the genes [ 1 ]. However, the origin of the genes and the evolutionary processes of such bacterial strains remain largely unknown. Ongoing comprehensive genome and metagenome analyses may provide some insights into these mysteries, and the genes for the degradation of xenobiotics can be used as probes to reveal novel mechanisms for the evolution of microorganisms. In addition, enzymes for the degradation of xenobiotics are good materials for studies on protein evolution, since generally they have promiscuous activities, and their properties change dramatically with a small number of mutations [ 2 ]. On the other hand, the importance of microbial consortia and symbiosis for the degradation of xenobiotics in the environment has also been suggested [ 3 ], and thus studies on xenobiotics degradation may provide some novel concepts in the field of microbial ecology.
This issue gathers 13 articles dealing with various aspects of the microbial degradation of xenobiotics. Four of them deal with the bacterial strains that degrade monocyclic phenolic compounds [ 4 ], polylactic acid [ 5 ], and naphthalene [ 6 ], and those that accumulate perfluorohexane sulfonate [ 7 ]. Two are dedicated to bacterial consortia degrading diesel [ 8 ] and dioxane [ 9 ]. Two focus on the enzymes for degradation of haloalkanes [ 10 ] and bisphenols [ 11 ]. Three articles are related to “indirect” factors that are necessary or important for the microbial degradation of xenobiotics, i.e., transcriptional regulation [ 12 ], transporters that are involved in the transport of xenobiotic compounds across the outer membrane [ 13 ], and mobile genetic elements [ 14 ]. The last two articles address metabolic engineering [ 15 ] and the bioreactors [ 16 ] necessary for practical application.
Acknowledgments
I would like to thank all authors who contributed their excellent papers to this Special Issue. I thank the reviewers for their help in improving the papers to the highest standard of quality. I am also grateful to all members of the Microorganisms Editorial Office for giving me this opportunity, and for their continuous support in managing and organizing this Special Issue.
Conflicts of Interest
The author declares no conflict of interest.
IMAGES
VIDEO
COMMENTS
The human gut microbiota makes key contributions to the metabolism of ingested compounds (xenobiotics), transforming hundreds of dietary components, industrial chemicals, and pharmaceuticals into metabolites with altered activities, toxicities, and lifetimes within the body. The chemistry of gut microbial xenobiotic metabolism is often distinct ...
Deciphering how gut microbial transformation of xenobiotics affects host health will require the integration of clinical studies with mechanistic experiments in model systems and organisms . These efforts will necessitate identifying microbial genes and/or metabolites that are reliable, diagnostic markers for activities of interest.
The understanding of how exogenous chemicals (xenobiotics) are metabolized, distributed, and eliminated is critical to determine the impact of the chemical and its metabolites to the (human) organism. This is part of the research and educational discipline ADMET (absorption, distribution, metabolism, elimination, and toxicity). Here, we review the work of Jan Commandeur and colleagues who have ...
4. Chemical Case Studies. In order to understand the mechanism of toxicity of chemicals, it is crucial to understand their metabolism. This is relevant for understanding safety issues with xenobiotics, drug toxicity through bioactivation into toxic metabolites, and drug efficacy. The following examples were worked on by Commandeur and his ...
Xenobiotics are encountered by humans on a daily basis and include drugs, environmental pollutants, cosmetics, and even components of the diet. ... Recent large-scale human population studies have illustrated how genetic and environmental differences can impact the metabolome. ... and ultimately the bioavailability, efficacy (in the case of ...
There is an increasing number of studies published on metabolomics-based POPs exposure both in vivo and in vitro. Table 1 presents selected studies with essential parameters, such as animal model utilized, exposure levels of xenobiotics, sample preparation conditions, and analytical instruments along with the columns used. In this review, we ...
Xenobiotics include plant components, pharmaceutical drugs, pesticides, cosmetic products, added food flavors, fragrances, etc. ... as in case of human hormones' uptake by fish in downstream of sewage treatment plant or chemical defenses used by some organisms against ... Studies of migration of pharmaceutical chemicals from soil to plants ...
Although several enzyme systems participate in phase I metabolism of xenobiotics, perhaps the most notable pathway in this scheme is the monooxygenation function catalyzed by the cytochrome P450s (CYPs; P450s). ... As is the case for the research on phase I biotransformation, research advances in the study of phase II metabolism saw explosive ...
foreign compounds (xenobiotics), including die-tary components, environmental pollutants, and pharmaceuticals. Such transformations were iden-tified as early as the 1950s in humans, animal models, fecal samples, and individual microbes. In these studies, changes in metabolism in the absence of microbes [i.e., germ-free (GF) animals]
Xenobiotic. A xenobiotic is a chemical substance found within an organism that is not naturally produced or expected to be present within the organism. It can also cover substances that are present in much higher concentrations than are usual. Natural compounds can also become xenobiotics if they are taken up by another organism, such as the ...
Current difficulties and limitations in xenobiotics-microbiota studies. Previous studies have evaluated the gut microbiota perturbation induced by a series of xenobiotics, but in most cases, we only demonstrated the correlation of xenobiotic exposure, gut microbiota dysbiosis, and disease outcomes, that unexposed and exposed subjects had ...
To date, very few studies have investigated the effect of feeding state on chemical-induced gut microbial metabolic dysregulations. Here, we set up an in vitro human gut microbiome and incorporated a metabolomics approach to investigate the effect of tetracycline (TET) at multiple doses (i.e., 10, 1, and 0.01 mg/L) on gut microbiome under the ...
The interactions of xenobiotics with the microbiota as a result of drug therapy or environmental exposures are of increasing interest to public health. ... Case studies provide examples of potential biomarkers that could serve as the basis for noninvasive diagnostic tests to identify disease and track its progression. When an association ...
Global environmental contamination with a complex mixture of xenobiotics has become a major environmental issue worldwide. Many xenobiotic compounds severely impact the environment due to their high toxicity, prolonged persistence, and limited biodegradability. Microbial-assisted degradation of xenobiotic compounds is considered to be the most effective and beneficial approach. Microorganisms ...
This study is the first to develop methods to define the metabolically active set of gut bacteria and demonstrate that xenobiotics shape the structure and physiology of these bacteria.
Global environmental contamination with a complex mixture of xenobiotics has become a major environmental issue worldwide. Many xenobiotic compounds severely impact the environment due to their high toxicity, prolonged persistence, and limited biodegradability. Microbial-assisted degradation of xeno …
Currently, environmental contamination with complex xenobiotic compounds has become a major global problem. Several recalcitrant xenobiotic compounds impact the environment mainly due to the high toxicity, prolonged persistence, and limited biodegradability of these compounds.
Xenobiotics are chemical substances not naturally produced or expected to be present within organisms. The term "xenobiotic" is usually used in the context of environmental pollutants to refer to synthetic compounds produced in large volumes for industrial, agricultural, and domestic use [2], [12].There is growing public concern over the wide range of xenobiotic compounds being introduced ...
From the past few. decades, microbial-assisted degradation (bioremediation) of xenobiotic pollutants has evolved as the most e ective, eco-friendly, and valuable approach. Microorganisms have ...
Chromatographic analysis of xenobiotics are used for separation and determination of compounds with similar chemical structures in the air, ground, in surface water, sludge, soil matrices, food and food products, and in human and veterinary health care. GC methods need compounds that are volatile or semi-volatile, such as toluene, xylene, and ...
chemical case studies by Commandeur and his colleagues that led to a better understanding of xenobiotic metabolism. 2. HALOGENATED ALKENES (WHERE IT ALL BEGAN) Jan Commandeur's Ph.D. thesis was entitled "Molecular mechanisms of chemically induced nephrotoxicity: Role of the mercapturic acid pathway in the bioactivation of halogenated ...
Thus, xenobiotic metabolism has great implication for chemical safety evaluation, which has become one of the central research areas in chemical toxicology. A plethora of analytical and in vitro methods are now available for investigating the metabolic fate of xenobiotics, especially by cytochrome P450 (CYP), at a high level of detail. However ...
Of 38,353 persons interviewed, 595 were found to have undiagnosed COPD or asthma and 508 underwent randomization: 253 were assigned to the intervention group and 255 to the usual-care group.
Clinical trials and investigations. Case study. VDEC is supporting a GBS vaccine to prevent newborn deaths. Antimicrobial resistance to Group B Streptococcus (GBS) antibiotics is growing. This ...
May 18, 2024, 11:00 AM UTC. By Aria Bendix. New research published this week lends credence to the more than 50,000 lawsuits against Johnson & Johnson that allege its talc-based baby powder caused ...
The company estimates a potential carbon footprint reduction of 34% from the project. In 2022, Pierre-Etienne Bindschedler, the president and third-generation owner of Soprema, set a goal to ...
27 Teams will examine asset and liability management . Washington, D.C. - Twenty-seven student teams from 21 colleges and universities across the nation have entered the 2024 CSBS Community Bank Case Study Competition.Each team has partnered with a local community bank to learn about the closures of Silicon Valley Bank, Signature Bank, and First Republic Bank, identify the case study bank ...
Predicting the biotransformation of xenobiotics is important in toxicology; however, as more compounds are synthesized than can be investigated experimentally, powerful computational methods are urgently needed to prescreen potentially useful candidates. Cytochrome P450 enzymes (P450s) are the major enzymes involved in xenobiotic metabolism, and many substances are bioactivated by P450s to ...
Using QuickSight and Redshift, Trustly estimates that its analytics solution generates insights four times faster than the previous one. "With the same team size, we can deliver more," says Padilha. "The solution increased our visibility, monitoring, availability, and resilience.". Since completing the project, Trustly has rapidly ...
Xenobiotics are released into the environment by human activities, and they often cause problems such as environmental pollution, since most such compounds cannot be readily degraded, and have harmful effects on human beings and the natural ecosystem. ... and thus studies on xenobiotics degradation may provide some novel concepts in the field ...